
Quantum chemical design of carbazole- and pyridoindole-based ambipolar host materials for blue phosphorescent OLEDs†
E. Varathanabd,
Dolly Vijayc and
V. Subramanian*abd
aChemical Laboratory, CSIR-Central Leather Research Institute, Adyar, Chennai-600 020, India. E-mail: subuchem@hotmail.com; subbu@clri.res.in; Fax: +91 44 24911589; Tel: +91 44 24411630
bAcademy of Scientific and Innovative Research (AcSIR), New Delhi 110025, India
cDepartment of Chemistry, University of Delhi, Delhi 110007, India
dCSIR-Network of Institutes for Solar Energy, India
First published on 1st August 2016
Abstract
Density functional theory has been employed to design thirty two host molecules for blue electrophosphorescence by incorporating dibenzofuran (DBF), dibenzothiophene (DBT), phenylcarbazole (Ph-Cbz), benzofuropyridine (DBF), benzothiopyridine (BTP) and phenyl pyridoindole (Ph-Cb1) subunits into N-phenyl carbazole (Ph-Cbz) and phenyl α-carboline (Ph-Cb1) core units. We have systematically investigated the role of linking positions of subunits into the core moiety and nature of core units on the electronic properties of the newly developed host materials. Results illustrate that substituting the subunits at the Ph-Cbz core may yield hosts with improved electronic properties when compared to the same subunit at the Ph-Cb1 core. The electronic properties are modulated efficiently through the nature and substituted positions (para and meta) of the subunits at the core units. Substitution of the subunits at the para-position of the Ph-Cbz core yields hosts with better charge injections. The same substitution at the meta-position results in better charge transport, higher triplet energy and lower singlet–triplet energy difference (ΔEST). Among the newly designed host molecules, 25, 26, 27, 28, 29, 30, 31 and 32 are found to be promising hosts molecules with a lower barrier for hole and electron injection, a clear charge-separated state, balanced charge transport for both hole and electron, and lower ΔEST values compared with an experimentally reported high potential host molecule (host 3).
1. Introduction
Recent studies have focused on organic light emitting diodes (OLEDs) using heavy transition-metal doped phosphorescence emitting materials (PhOLEDs), because these materials may allow 100% internal quantum efficiency (IQE) by means of facile spin–orbit coupling.1–3 It is well known that host molecules are an important component of PhOLEDs. The phosphorescent emitters are usually doped within suitable host materials to reduce concentration quenching and triplet–triplet annihilation.4 The development of host materials for blue PhOLEDs is particularly challenging because host molecules are required to have both high triplet energy (ET > 2.8 eV) and good charge-carrier transport properties. To obtain efficient blue PhOLEDs, the performance of the host molecule needs to fulfill the following criteria: (i) the triplet energy (ET) of the host molecule should be higher than that of the phosphorescent guest emitter, to prevent the back energy transfer from guest to host and facilitate the exothermic energy transfer from host to guest,5,6 (ii) to achieve low operating voltage, the Highest Occupied Molecular Orbital (HOMO, (ionization potential (IP))) and Lowest Unoccupied Molecular Orbital (LUMO, (electron affinity (EA))) levels of the host molecule should be appropriately aligned with respect to the neighboring hole transport layer (HTL) and electron transport layer (ETL), (iii) the host materials should have good charge-carrier transport properties, and (iv) high thermal stability and film forming ability. These limitations make the design and development of efficient host materials for blue PhOLEDs particularly challenging compared to red and green. Over the past few years, several efforts have been devoted to develop efficient host materials for blue PhOLEDs. Carbazole derivatives are among the most favorable materials in this context owing to their rigid molecular frame, high ET level, good thermal stability, and excellent hole transporting properties.7–15 Among the various designing strategies for the development of host materials, bipolar/ambipolar host materials have been the subject of interest because these materials can transport both hole and electron in a well-balanced manner that could extend the emitting zone in an emitting layer.16,17 Ambipolar host materials are composed of an electron-donating moiety which is capable of mediating hole injection and transportation and an electron-withdrawing moiety which facilitates electron injection and transportation. A large number of ambipolar host molecules have been reported in the literature.8,18–29 For example, Kido and co-workers have developed host materials with carbazole and pyridine-based terphenyl like subunits, with external quantum efficiency (EQE) of 24% for blue emiters.23 In addition several host molecules have been designed using quantum chemical calculations.30–39 In particular, Brédas and coworkers have studied a number of ambipolar host molecules using quantum chemical methods. They have reported that in the designing of ambipolar host molecules the inductive and mesomeric effect of each unit plays an important role.7,40–42Recent investigations have demonstrated that the pyridoindole (α-carboline) moiety would be an effective building block to construct an efficient ambipolar host molecule for blue PhOLEDs.43–50 In these studies several host molecules which engage α-carboline unit have been developed and the corresponding blue PhOLEDs device have exhibited significantly improved device efficiency. Particularly, Lee and co-workers have reported a number of pyridoindole derivatives as the host materials for blue PhOLEDs.44–46,51–60 Recently, the same group46 synthesized a series of host molecules including 1, 2, and 3 (Scheme 1) by introducing dibenzofuran (DBF), dibenzothiophene (DBT), and 9-phenylcarbazole (Ph-CBz) substituted groups into α-carboline core with the aim to develop efficient host molecules for blue PhOLEDs. These authors demonstrated that the device, in which 3 is used as the host along with Flrpic as the blue emitter, has considerably high external quantum efficiency of 29.6%. These authors also reported that substituting Ph-CBz unit in the meta-position of Ph-Cb1 core provided the appropriate energy levels and charge-transport properties of the pyridoindole-derived host when compared to the substitution by DBF or DBT. The same group also reported 24% EQE in blue PhOLEDs using 1 (see Scheme 1) as a host material and Flrpic phosphorescent dopant. This host has a DBF unit attached at the meta-position of the Ph-Cb1 core. Interestingly, when the central core unit of host 1 is replaced by Ph-CbZ core, the resulting host 13 has lower device performance (EQE = 19.6%) compared to host 1.56 These observations highlight that variation in the core structure of the host molecules can change the quantum efficiency of the device. The electronic properties of the host molecules can alternatively be altered by changing the subunits. For example, Lee and co-workers,59 have successfully synthesized a 6-position modified benzofuro[2,3-b]pyridine (BFP) derivative (host 16 in Scheme 1). This host has BFP unit attached at the meta-position of the Ph-CBz core and has better EQE (∼24.3% EQE with FIrpic) in contrast to host 13. In our previous study, we have observed that the position of the substituents is also an important factor that should be considered while designing host molecules.39 The preceding discussion clearly reveals that both substituted core and substituting units (subunits) play an important role in determining the efficiency of the host molecules. Therefore, a comprehensive systematic study on the effect of core and substituting moieties on the electronic properties of host molecules is necessary.
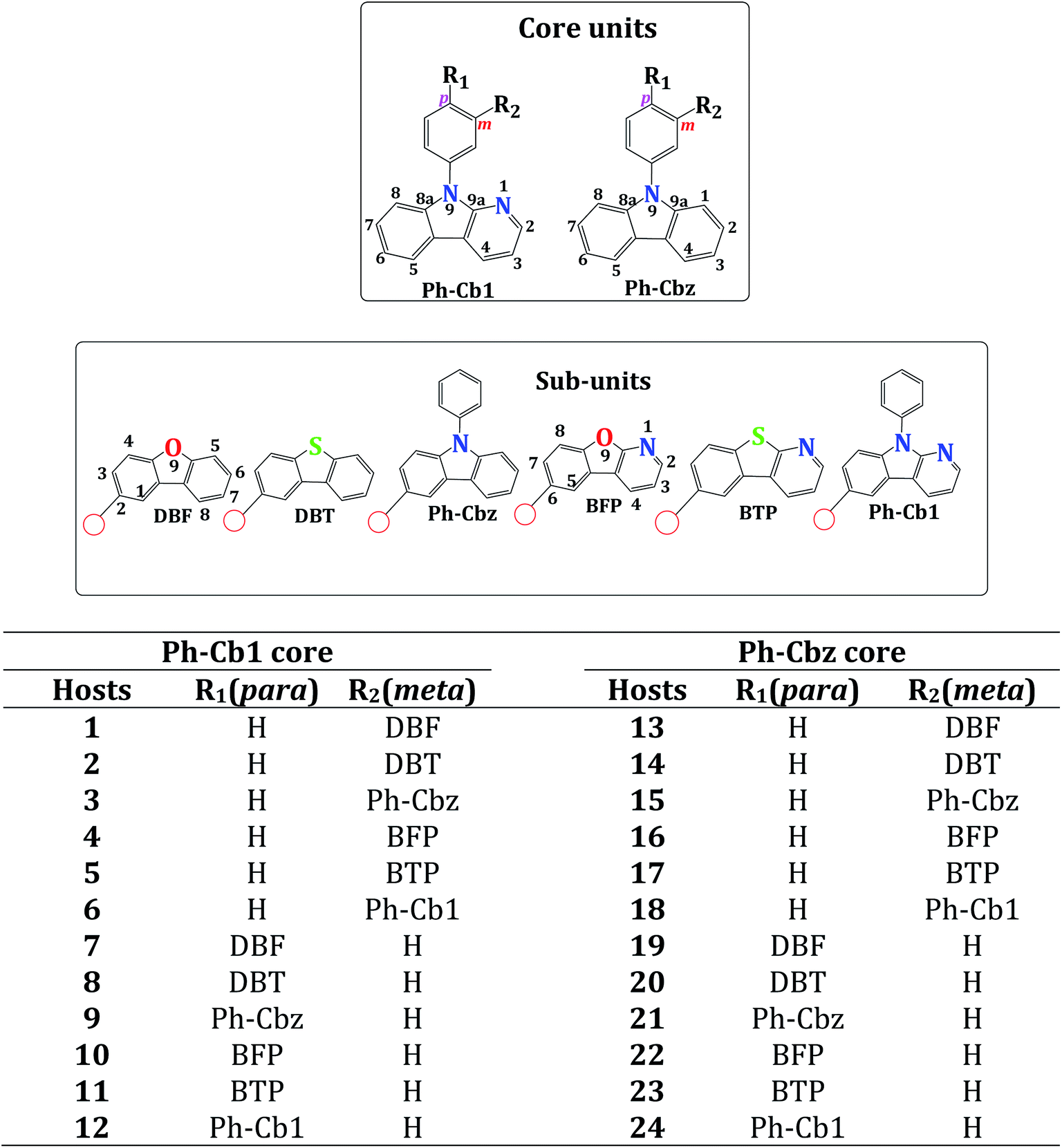 | ||
| Scheme 1 Chemical structure of core and subunits used to design new host molecules. | ||
In this work, we have considered phenyl carbazole (Ph-Cbz) and phenyl α-carboline (Ph-Cb1) as the core unit and dibenzofuran (DBF), dibenzothiophene (DBT), phenylcarbazole (Ph-Cbz), benzofuropyridine (DBF), benzothiopyridine (BTP) and phenyl pyridoindole (Ph-Cb1) as a subunits to construct the host molecules. The main impetus of the study is to analyze the variation in the electronic properties when different subunits are introduced into the Ph-Cbz and Ph-Cb1 core, and which core unit is more suitable to obtain an ambipolar host material with improved electronic properties with reference to already reported hosts (1, 3, 13 and 16). We would also like to investigate how the nature and the linking positions (para (p) and meta (m)) of subunits would influence the electronic properties such as ET, HOMO and LUMO, ΔEST, charge injection, and charge transport properties.
To elucidate the structure–property relationship, density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations have been performed to gain an in-depth understanding of the geometries, and electronic properties by considering a twenty-four host molecules which are designed by using two different core units substituted by six different subunits. It is expected that the predicted structure–property relationships would give clear insights into the main issues regarding the design and development of carbazole and carboline based host molecules for future development of the branch of carboline based host for blue PhOLEDs.
2. Computational details
The ground state geometries were optimized by using density functional theory (DFT) B3LYP functional.61,62 The structures were characterized as minima by employing frequency calculations which yielded all positive frequencies. The choice of the functional is based on the benchmark study. In this regard, we carried out calculations on nine molecules (hosts 1, 2, 3, 13 and 16; and reference HTL (TAPC), ETL (TmPyPB), emissive (FIrpic) and host (mCP)) for which experimental HOMO and LUMO data were available. For the benchmark calculations, we optimized the nine molecules using an array of functionals viz. BP86, O3LYP, B3LYP, PBE0, M06, BMK, BH&HLYP, M06-2X, CAM-B3LYP, ω-B97XD, LC-ωPBE and M06-HF (0%, 20%, 20%, 25%, 27%, 42%, 50%, 54%, 20–65% (20% short range (SR) and 65% long range (LR)), 15.77–100%, 0–100% and 100% HF exchange, respectively) in conjugation with the 6-31+G* basis set. The HOMO–LUMO values reported in Table S1 (ESI†) clearly indicate that the HOMO energy is linearly related to the fraction of the HF exchange in the functional, and that the PBE0, M06 and B3LYP functional exhibit a smaller deviation from the experiment values than the other functionals. On the other hand, calculated LUMO energy values by various functionals are substantially higher in energy when compared with experimentally obtained values because the virtual orbitals are generally more difficult to describe theoretically than the occupied orbitals. The results point out that the B3LYP functional can predict the both HOMO and LUMO values close to the experimental values unlike other functional which may predict either HOMO or LUMO with appreciable accuracy. These findings substantiate that B3LYP functional is suitable to predict the electronic properties of these molecular systems. Furthermore, we have also calculated the IP and EA values of the nine reference molecules using optimized ionic and neutral geometries. The calculated values are displayed in Table S2 (ESI).† The experimental trends in HOMO, LUMO, IP and EA values are reproduced by the calculated values (Fig. S1 and S2 in ESI†).A host of literature can be found which indicate a strong correlation between the energies of vertical excited states obtained via the TD-DFT approach and the percentage of the HF-like exchange employed.42,63,64 In order to analyze how the variation in the choice of functional can alter the vertical S1/T1 excitation energies, we carried out vertical excited state calculations on five experimentally reported host molecules (hosts 1, 2, 3, 13 and 16) with functionals like B3LYP, CAM-B3LYP, M06, M06-2X, PBE0,ωB97X and LCω-PBE in conjunction with the 6-31G* basis set (details are provided in the ESI†). The B3LYP/6-31G* optimized geometries were used for benchmarking various functionals. Among the various functionals considered in this study, B3LYP and M06-2X exhibit good agreement with experimental data (Fig. S3†). Thus, we have restricted all the analysis to the B3LYP functional. The adiabatic ET energy were calculated by means of the ΔSCF65–67 method by using the optimized geometries of T1 and S0 states. The spin-unrestricted B3LYP (UB3LYP) was adopted to optimize the structures of stable triplet (T1) states in conjugation with the 6-31G* basis set. The stable geometries of S1 states were optimized via a time-dependent density functional theory (TD-DFT) method with B3LYP/6-31G* level of theory. To further investigate the effect of basis sets on adiabatic ES and ET energies, we calculated the adiabatic ES and ET energies with 6-31G* and 6-31+G* basis set and the results are shown in Fig. S4 (ESI).† As shown in Fig. S4,† both basis sets predicted the similar trends of the ΔEST values for five experimentally reported host materials. Hence, for all the adiabatic ES and ET values were calculated using 6-31G* basis set. In the case of the triplet energies, zero point vibrational energy (ZPVE) correction was included. To gain further insights into the nature of the triplet state, natural transition orbital analysis (NTO)68 was performed based on the TD-DFT approach, using the optimized geometries of the triplet state. All calculations were carried out using the Gaussian 09 package.69
We choose N,N′-dicarbazolyl-3,5-benzene (mCP) as a reference host molecule for blue electrophosphorescence and di[4-(N,N-ditolyl-amino)-phenyl]cyclohexane (TAPC) and 1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB) as reference hole and electron transport materials, respectively. Iridium(III) bis((4,6-difuorophenyl)pyridinato-N,C2′)picolinate (FIrpic) was taken as the reference blue emitting material. The geometry optimization of the reference emitter was carried out at B3LYP level with LANL2DZ basis set for the iridium atom and 6-31+G* for the rest of the atoms.
2.1 Carrier mobility
Previous studies concluded that the charge mobility of organic molecules can be well described by the incoherent hopping model.70–72 In this model, the charge transport is viewed as a charge exchange reaction.| Mn + Mi → Mi + Mn | (1) |
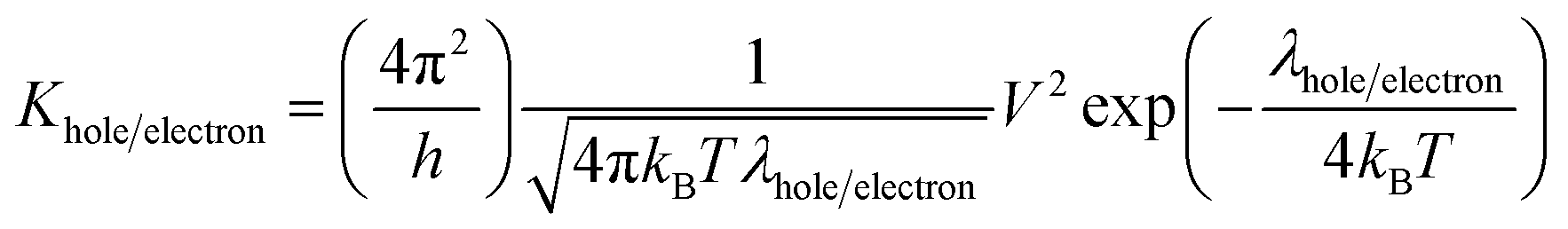 | (2) |
| λ+ = λ1 + λ2 | (3) |
| λ− = λ3 + λ4 | (4) |
| λ1 = E+(M0) − E+(M+); λ1 = E0(M+) − E0(M0) |
| λ3 = E−(M0) − E−(M+); λ1 = E0(M+) − E0(M0) |
The electronic coupling matrix element (Vab) represents the strength of electronic coupling between the two adjacent neighboring molecules a and b. Though the molecular stacking can generate a large number of conformations, being with extended π-conjugation, the intermolecular stacking is expected to be dominantly influenced by the π–π interactions.71 Indeed, face to face π-stacking with a large orbital overlap facilitates the enhanced electronic coupling between the two adjacent molecules and consequently aids the inter-chain charge transport of an organic molecule. It is evident from previous reports, M06-2X functional yields reliable results for non-covalent interactions and dispersion-dominated systems.76–80 Hence, M06-2X is well suited for the optimization of dimer. To calculate the transfer integral (Vab), we used M06-2X/6-31G* level of theory to optimize the dimer structure and single point calculations were performed at M06-2X/6-31++G** level of theory using optimized geometries as obtained from M06-2X/6-31G* method to calculate the transfer integral. The optimized structures of the host dimers are shown in Fig. S5 (ESI).†
In this study, Vab was calculated based on Koopmans theorem (KT)81 by assuming that the interacting molecules are identical, symmetrically equivalent and also have the same site energies.71 According to KT, the transfer integrals are estimated as half the splitting of the HOMO levels (for holes) and LUMO levels (for electrons) induced by the interaction of monomer orbitals (HOMOs and LUMOs) in a dimer. It is given as:
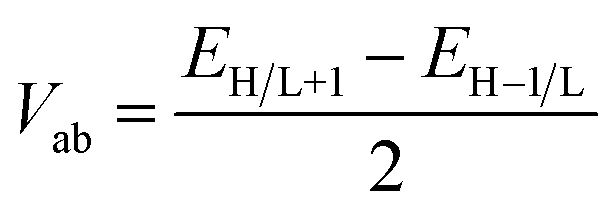 | (5) |
However, application of KT theorem may fail, as pointed out by Brédas et al.84 The inclusion of polarization (but often ignored) is important for organic molecules due to considerable impact of polarization on the site energies. In the same study the authors also emphasized that a proper orthogonalization of the initial and final states is an important to obtain correct values of electronic coupling. The optimized dimer structures of all the host materials in this study exhibited nearly co-facially stacked structures (Fig. S5†). Hence, the site energy correction was not considered. In this study the hopping mobility can be estimated by using the Einstein relation.
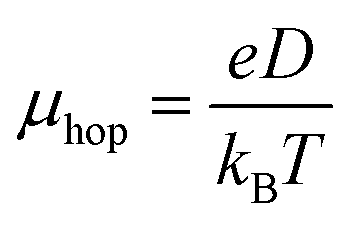 | (6) |
3. Results and discussions
In the present work, based on the substitution of subunits on the Ph-Cb1 or Ph-Cbz core, 24 unique host molecules have been designed (Scheme 1) and these are classified as Ph-Cb1-based and Ph-Cbz-based host molecules in the remaining part of the text.3.1 Electronic structure of Ph-Cb1-based host molecules
We start by explaining the electronic properties of the series of Ph-Cb1 based host molecules. The substitution of DBF, DBT, Ph-Cbz, DBF, BTP and Ph-Cb1 at the meta position of Ph-Cb1 leads to hosts 1, 2, 3, 4, 5, and 6, respectively. The substitution of respective subunits at the para position of Ph-Cb1 yields hosts 7, 8, 9, 10, 11, and 12. Totally, twelve host molecules have been designed. The HOMO and LUMO energies are important parameters in designing host materials for OLED applications. A good host material should have optimum HOMO and LUMO energies to reduce the charge injection barrier from the neighboring layers (HTL/ETL). As shown earlier study,40 for the efficient hole transport from HTL to host molecule, HOMO level of the host should be close to that of HOMO level of the HTL. Moreover, HOMO level of the host should be in between that of HOMO level of HTL and emitter. Similarly, for electron transport from the ETL to host molecule, the LUMO level of the host molecules should be close to LUMO of ETL. To assess energy barriers for charge injection from neighboring layers, the HOMO and LUMO of the designed host materials are presented in Fig. 1 (also listed in ESI, Table S2†) along with those of HTL, ETL and emissive layer. The calculated HOMO energies for hosts 1–12 vary from −5.50 to −5.96 eV. It can be observed that the nature and position of the substituents at the Ph-Cb1 core determine the electronic properties. It can be clearly seen from Fig. 1 that the HOMO values of all the designed host molecules (expect hosts 4 and 5) range from −5.50 to −5.89 eV which are in between HTL (−4.91 eV) and emissive layer (−5.91 eV). Hence, these host molecules can efficiently mediate the hole transport between HTL and emissive layer. However, close analysis reveals that the hosts 4 and 5 are different from other host molecules. The HOMO energy level is not energetically in alignment with that of emissive layer. If host 4 and 5 are used as the host materials, the hole transfer from HTL (TAPC) to emissive (FIrpic) layer would require a higher turn-on voltage. Among the designed host molecules, the HOMO levels of 3 (−5.56 eV), 9 (−5.50 eV) and 12 (−5.66 eV) are high which imply that these molecules have the lowest hole injection barrier. In addition, the HOMO energies of the hosts 3, 6 and 9 are higher in energy than that of reference host mCP (−5.71 eV) which is a widely used hole transport materials in PhOLEDs. Thus, these host molecules (3, 9 and 12) may have better hole transport ability than mCP. As pointed out by Brédas et al.,85 FMO energy levels (HOMO and LUMO) derived from experimental measurement related to adiabatic ionization potentials (IPs) and electron affinities (EAs) energies. In order to get energy barriers for charge injection from neighboring layers, it is necessary to compare the IPs and EAs of the corresponding molecules. It is evident from the previous reports that the efficient hole injection from HTL to the emitter occurs only if the IP value of the host molecule is in between those of the HTL and emitters.38,40–42 Similarly, for facile electron injection from the ETL to host, the electron affinity difference between the ETL and host should be low. The calculated IP/EA values of the designed host molecules are collected in Table 1 along with those of TAPC, TmPyPB, and FIrpic. The calculated IP values (6.41–6.94 eV) of the Ph-Cb1-based hosts (expect hosts 1, 4 and 5) lie in between those of TAPC (IP = 5.80 eV) and FIrpic (IP = 6.96 eV). Hence, these hosts can potentially mediate holes from HTL to the host matrix and to the guest emitter. In the case of hosts 1, 4 and 5, the calculated IP values are predicted to be 7.01, 7.13 and 7.06 eV respectively, which are higher than that of TAPC and FIrpic. This evidence clearly shows that these three hosts exhibit a large barrier for hole injection from HTL to the host matrix and to the guest emitter. Among the host materials, the hosts 3, 9 and 12 exhibits lower IP values when compared with reference host materials (mCP, IP = 6.80 eV). Inspection of calculated IP values with HOMO energies yields the similar picture. For facile electron injection, the LUMO of the hosts should be in alignment with that of the ETL. The calculated LUMO energies for hosts 1–12, vary from −1.35 to −1.75 eV. It is possible to note that these values depend on the nature and position of the subunits at the Ph-Cb1 core. The LUMO energies of the hosts fluctuate around that of ETL (−1.52 eV). The comparison EA values (Table S2†) provides similar findings. The calculated EA values for the isolated host materials are found to be in the range of 0.29–0.63 eV, which are close to that of TmPyPB (EA = 0.68 eV). These findings suggest that, these host materials are well positioned with respect to ETL, which leads to low barrier for electron injection from ETL to host molecules. The noteworthy points that emerge from the preceding discussion are as follows: (i) electron injection barrier is not affected by the position of subunits at the Ph-Cb1 core, whereas hole injection barrier is mainly influenced by the position of the subunits at the Ph-Cb1. (ii) The substitution at p-position yields better hole injections barrier when compared to m-position. | ||
| Fig. 1 HOMO and LUMO energy levels (in eV) and HOMO/LUMO energy gap (Eg) of Ph-Cb1-based hosts and reference HTL (TAPC), ETL (TmPyPB), and emitter (FIrpic) as obtained at the B3LYP/6-31+G* level. | ||
| Hosts | Cb1 core unit | Cbz core unit | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IP | EA | λh | λe | Δλ | ETa | E0Ta | ES | ΔEST | Hosts | IP | EA | λh | λe | Δλ | ETa | E0Ta | ES | ΔEST | |
| a ET and E0T denote the adiabatic triplet energy without and with zero-point vibrational (ZPVE) energy correction. | |||||||||||||||||||
| 1 | 7.01 | 0.38 | 0.20 | 0.12 | 0.08 | 3.06 | 2.89 | 3.57 | 0.68 | 13 | 6.90 | 0.31 | 0.12 | 0.19 | 0.07 | 3.17 | 2.98 | 3.49 | 0.51 |
| 2 | 6.94 | 0.39 | 0.15 | 0.13 | 0.02 | 2.96 | 2.82 | 3.57 | 0.75 | 14 | 6.85 | 0.36 | 0.09 | 0.37 | 0.28 | 2.94 | 2.81 | 3.42 | 0.61 |
| 3 | 6.61 | 0.29 | 0.18 | 0.10 | 0.08 | 3.15 | 2.98 | 3.52 | 0.54 | 15 | 6.59 | 0.24 | 0.10 | 0.31 | 0.21 | 3.15 | 2.99 | 3.75 | 0.76 |
| 4 | 7.13 | 0.59 | 0.21 | 0.12 | 0.09 | 3.05 | 2.90 | 3.58 | 0.68 | 16 | 6.99 | 0.55 | 0.12 | 0.17 | 0.05 | 3.06 | 2.90 | 3.38 | 0.48 |
| 5 | 7.06 | 0.59 | 0.16 | 0.13 | 0.03 | 3.08 | 2.91 | 3.58 | 0.67 | 17 | 6.95 | 0.54 | 0.10 | 0.18 | 0.08 | 3.09 | 2.92 | 3.40 | 0.48 |
| 6 | 6.75 | 0.47 | 0.19 | 0.15 | 0.04 | 3.06 | 2.87 | 3.46 | 0.59 | 18 | 6.72 | 0.41 | 0.11 | 0.19 | 0.08 | 3.07 | 2.89 | 3.47 | 0.58 |
| 7 | 6.79 | 0.42 | 0.35 | 0.12 | 0.23 | 3.05 | 2.89 | 3.38 | 0.49 | 19 | 6.74 | 0.35 | 0.23 | 0.22 | 0.01 | 2.88 | 2.76 | 3.44 | 0.68 |
| 8 | 6.73 | 0.43 | 0.30 | 0.13 | 0.17 | 2.84 | 2.72 | 3.38 | 0.66 | 20 | 6.69 | 0.41 | 0.20 | 0.39 | 0.19 | 2.82 | 2.70 | 3.38 | 0.68 |
| 9 | 6.41 | 0.32 | 0.32 | 0.11 | 0.21 | 2.84 | 2.71 | 3.24 | 0.53 | 21 | 6.41 | 0.28 | 0.24 | 0.23 | 0.01 | 3.14 | 2.98 | 3.53 | 0.55 |
| 10 | 6.90 | 0.63 | 0.36 | 0.12 | 0.24 | 3.05 | 2.89 | 3.53 | 0.64 | 22 | 6.84 | 0.56 | 0.23 | 0.18 | 0.05 | 2.88 | 2.75 | 3.39 | 0.64 |
| 11 | 6.84 | 0.63 | 0.32 | 0.14 | 0.18 | 2.84 | 2.72 | 3.49 | 0.77 | 23 | 6.79 | 0.57 | 0.21 | 0.18 | 0.03 | 2.81 | 2.70 | 3.39 | 0.69 |
| 12 | 6.55 | 0.51 | 0.34 | 0.15 | 0.19 | 2.84 | 2.72 | 3.38 | 0.66 | 24 | 6.53 | 0.44 | 0.25 | 0.18 | 0.07 | 2.82 | 2.70 | 3.36 | 0.66 |
| mCP | 6.80 | 0.15 | 0.06 | 0.12 | 0.06 | 3.17 | 2.98 | 2.15 | 0.83 | ||||||||||
| FIrpic | 6.96 | 1.06 | — | — | — | 2.71 | 2.59 | — | — | ||||||||||
| TAPC | 5.80 | 0.11 | — | — | — | — | — | — | — | ||||||||||
| TmPyPB | 7.41 | 0.68 | — | — | — | — | — | — | — | ||||||||||
To understand the evolution of the HOMO and LUMO energies upon substitution, we have systematically analyzed the HOMO and LUMO values of the Ph-Cb1 based hosts and their core unit. A comparison of the HOMO–LUMO (H–L) gap of Ph-Cb1 and the designed hosts (Fig. 1) indicates the presence of profound variations in both the HOMO and LUMO energy levels. The HOMO (IP) values are stabilized by 0.01–0.38 eV (0.11–0.83 eV) and the stabilization of LUMO (EA) values range from 0.01–0.33 eV (0.23–0.54 eV). As expected, the electron donating groups could destabilize the HOMO levels with respect to electron withdrawing moieties.33 Hence, the hosts 3 and 9 exhibit more destabilized (0.32–0.38 eV) HOMO energy levels when compared to other host materials. It is also clear from the electron density distributions of HOMO and LUMO levels (Fig. S6†) that the electron density of HOMO levels of the hosts 3 and 9 are localized on the Ph-Cbz unit, whereas, for other hosts the electron density of HOMO levels are localized on the Ph-Cb1 core. In the case of LUMO, the more electron accepting group could stabilize the LUMO levels. The LUMO levels of the isolated subunits are presented in ESI (Table S7).† It can be seen from Table S3† that, both BFP (−1.71 eV) and BTP (−1.68 eV) exhibit stabilized LUMO energy levels when compared to other subunits and core. Thus, the LUMO levels of hosts 4, 5, 10 and 11 are more stabilized with reference to other host molecules. It is also reinforced by the presence of electron density distributions of LUMO levels on the BFP and BTP units. These findings clearly reveal that the substitution of BFP and BTP units at the Ph-Cb1 core could reduce the electron injection barrier.
To explore the influence of different positions (p and m) of the substitution on the HOMO (IP) and LUMO (EA) energy levels of the designed hosts, we have compared the hosts 1, 2, 3, 4, 5, and 6 with 7, 8, 9, 10, 11, and 12, respectively. It can be noticed from Table 1 and Fig. 1 that, p-substituted hosts (7, 8, 9, 10, 11, and 12) have destabilized HOMO (IP) energy levels in contrast to that of meta-substituted hosts (1, 2, 3, 4, 5, and 6). The difference in the HOMO (IP) energies of the hosts 7, 8, 9, 10, 11, and 12 with respect to 1, 2, 3, 4, 5, and 6 is 0.09, 0.12, 0.06, 0.08, 0.09 and 0.11 eV (0.22, 0.20, 0.19, 0.22, 0.22 and 0.21 eV), respectively, which indicate that p-substitution may improve the hole injection barrier. On the other hand, the variation in the LUMO (EA) energy levels is less sensitive to the position of substitution. The maximum variation of LUMO energies between para and meta-substituted hosts ranges from 0.01–0.02 eV (0.04–0.05 eV).
In order to analyze why substitution of subunits at the para-position of the Ph-Cb1 core destabilizes HOMO energy levels when compared to meta-position, the molecular orbital (MO) distributions of the individual core unit and host molecules were analyzed. The HOMO of Ph-Cb1 core is localized on the Cbz unit with a node on the C2 and C7, C12 and C14 carbon atoms (Scheme 1 and Fig. S7†). Also, the HOMO is characterized by high electron density on the amine nitrogen (N9) which suggests its electron-donating nature. On the other hand, LUMO of Ph-Cb1 core is concentrated on the Cbz unit with a node present on the C3 and C6 carbon atoms and node on the N9 nitrogen atom. As pointed out by Bréads et al., if the linking atoms contribute significantly to MO, the interaction between the subunits for that MO is significant.42 As we can observe from ESI (Fig. S6†) that all the p-substituted hosts molecules have a significant MO interactions between the core and subunits, which extends the electron density distribution of HOMO on the entire molecule. Consequently, anti-bonding interaction between the HOMOs of the core and subunits leads to the destabilization HOMOs in para-substituted hosts when compared to meta-substituted hosts. Irrespective of the positions of substitution, electron density distributions of LUMO of both para- and meta-substituted hosts are concentrated on the same unit. As a result, the LUMO energy level does not vary significantly upon changing the position (from m to p position) of the subunits at the Ph-Cb1 core.
It is also interesting to analysis the role of steric effect in the meta-substituted systems. To unravel steric effect, the deviation in torsion angle of host molecules from the unsubstituted core units in the ground state was calculated. The results are displayed in ESI (Table S4).† The maximum deviation in the torsion angle in Ph-Cb1 based host molecules is within 3°. Hence, the steric effect of meta-substituents does not change the conjugation between the core and substituents.
It is evident from the previous report86 that a clear spatial separation between the HOMO and LUMO levels of the host materials is necessary for the facile hole and electron transfer and to prevent back energy transfer. The same feature is due to the ambipolar charge-transport character of host molecules.42 The frontier molecular orbital (FMO) distributions and compositions of the Ph-Cb1-based hosts are presented in ESI (Table S5 and Fig. S6).† It is evident from these results that, clear charge separation can be seen in the hosts 3, 4, 5, 10, and 11. In host 3, the HOMO is localized on the Ph-Cbz (86.40%) unit, due to strong electron donating nature of the Ph-Cbz unit over Ph-Cb1. On the other hand, LUMO is only concentrated on the Cb1 core (99.81%), due to electron-accepting nature of pyridine unit in the pyridoindole moiety. In hosts 4, 5, 10 and 11, the HOMO is delocalized over the Ph-Cb1 (98.43–81.03%) core due to the presence of electron donating indole unit in Ph-Cb1 core. In these systems, LUMO is localized on BFP (∼94%) and BTP (∼94%), because of BFP and BTP units are more electron acceptor than Ph-Cb1 unit. In case of hosts 1, 2, and 6, both HOMO and LUMO are localized on the Ph-Cb1 core moiety. Although, LUMO levels of 7, 8, 9 and 12, are completely localized on the Cb1 (93.77–98.98%) core, the HOMO is delocalized on both core and subunits.
A potential host molecule should have good charge transport rate and balanced carrier transport. The reorganization energy (λ) and the intermolecular transfer integral are the two key parameters in determining the charge transfer rate. In order to evaluate the charge mobility, the λ values of the hosts were evaluated from the hole and electron relaxation energies (Table S2†). The reorganization energy is an important factor that governs the mobility of charge carriers (hole and electron). The low reorganization energy is associated with higher transport rate of the carrier. The calculated reorganization energies for hole (λh) and electron (λe) transport of the Ph-Cb1-based host molecules are listed in Table 1. It can be observed from the results that for all the hosts molecules, the reorganization energy for hole transport (λh) is higher than that for electron transport (λe). These findings suggest that the electron transport performance of these host molecules is more favorable than hole transport ability. The calculated λe values of the meta-substituted host molecules are lower (0.10–0.15 eV). The predicted λh values are marginally higher which vary from 0.15–0.21 eV. These results indicate that Ph-Cb1 is excellent electron transport building unit. Similarly, para-substituted host molecules also exhibit the same trend. The calculated λe values ranges from 0.11–0.15 eV which are comparable to meta-substituted molecules. The λh values of para-substituted host molecules are appreciably higher than meta-substituted (0.30–0.36 eV). It is possible note from these values that hole transfer rate of meta-substituted hosts are more favorable than those of para-substituted systems. Scrutiny of λh and λe energies of para- and meta-substituted hosts elicit that a balanced charge transport is highly feasible in meta-substituted hosts (1, 2, 3, 4, 5 and 6) as the reorganization energies for λh and λe are comparable. The Δλ values range from 0.02 to 0.09 eV which point out that these molecules may act as good ambipolar host materials.30 It can be noticed that linking topology has more impact on the hole reorganization energy in contrast to that of electron.
In order to obtain a rational explanation for the balanced charge transport in meta-substituted hosts in contrast to para-substituted systems, the electron density distributions of MO of the host molecules were derived from the calculations. It can be noted that except host 3, the HOMO energy levels of all other meta-substituted hosts are localized on the Ph-Cb1 unit. Hence, λh values are close to that of Ph-Cb1 unit (λh = 0.18 eV), which indicate that the Ph-Cb1 can act as a hole transporting moiety. On the other hand, in para-susbtuted hosts, HOMO energy levels are delocalized on both core and subunits, which elicit that core and subunits play the roles of both hole and electron transport units. Thus it is possible to expect higher λh values for para-substituted molecules when compared to meta-substituted host materials. However, the electron transporting units in all the host molecules are Ph-Cb1, BFP and BTP as the LUMO distribution entirely concentrated on these units. Hence, the λe energies are almost same for all the host molecules. The above-mentioned findings point out that (iii) the linking topology has a significant effect on the hole transport properties. (iv) Particularly, the introduction of subunits at meta-position significantly enhances the hole transporting ability of the host materials.
The calculated electronic coupling (V), inner reorganization energy (λ), hole transport rates (k), average intermolecular distance between two dimers (d), and maximum rate of charge hopping (K), and drift mobility (μ) for hole and electron of designed host materials are listed in Table 2. As can be seen from Table 2, meta-substituted host materials exhibit more hole drift mobility (0.38 to 6.58 cm2 V−1 s−1) compared to para-substituted hosts (0.21 to 11.79 cm2 V−1 s−1), which indicate that para-substituted hosts have higher hole mobility than meta-substituted hosts.87 The calculated electron mobility for meta-substituted hosts, varies from 4.77 to 18.01 cm2 V−1 s−1, and for para-substituted hosts, it ranges from 0.01 to 5.74 cm2 V−1 s−1. For all the host materials the calculated hole and electron mobility values substantially higher than those of experimentally reported values owing to the fact that these values depend on several external factors, including device fabrication condition, material processes, and disorder and impurities present in the organic film.
| Hosts | d (Å) | V (eV) | K (s−1) × 10−14 | μ (cm2 V−1 s−1) | |||
|---|---|---|---|---|---|---|---|
| Hole | Electron | Hole | Electron | Hole | Electron | ||
| 1 | 3.46 | 0.10 | 0.10 | 0.54 | 1.51 | 2.50 | 7.03 |
| 2 | 3.42 | 0.04 | 0.12 | 0.14 | 1.89 | 0.63 | 8.62 |
| 3 | 3.53 | 0.14 | 0.07 | 1.35 | 0.98 | 6.53 | 4.77 |
| 4 | 3.66 | 0.06 | 0.09 | 0.17 | 1.22 | 0.89 | 6.37 |
| 5 | 3.49 | 0.03 | 0.17 | 0.08 | 3.79 | 0.38 | 18.01 |
| 6 | 3.52 | 0.15 | 0.17 | 1.36 | 2.91 | 6.58 | 14.04 |
| 7 | 3.33 | 0.04 | 0.01 | 0.01 | 0.02 | 0.06 | 0.07 |
| 8 | 3.55 | 0.12 | 0.01 | 0.24 | 0.01 | 1.17 | 0.01 |
| 9 | 3.61 | 0.42 | 0.24 | 2.32 | 0.99 | 11.79 | 5.74 |
| 10 | 3.45 | 0.16 | 0.08 | 0.22 | 0.97 | 0.99 | 4.47 |
| 11 | 3.36 | 0.06 | 0.10 | 0.05 | 1.15 | 0.21 | 5.05 |
| 12 | 3.57 | 0.45 | 0.01 | 0.15 | 0.01 | 0.70 | 0.45 |
| 13 | 3.53 | 0.02 | 0.20 | 0.06 | 2.42 | 0.29 | 11.76 |
| 14 | 3.59 | 0.06 | 0.04 | 0.84 | 0.01 | 4.21 | 0.06 |
| 15 | 3.66 | 0.09 | 0.10 | 1.63 | 0.15 | 8.48 | 0.77 |
| 16 | 3.33 | 0.06 | 0.01 | 0.54 | 0.01 | 2.30 | 0.03 |
| 17 | 3.50 | 0.11 | 0.00 | 2.43 | 0.01 | 11.58 | 0.03 |
| 18 | 3.79 | 0.04 | 0.02 | 0.29 | 0.02 | 1.55 | 0.14 |
| 19 | 3.63 | 0.01 | 0.23 | 0.01 | 0.22 | 0.03 | 11.42 |
| 20 | 3.60 | 0.12 | 0.00 | 0.77 | 0.01 | 4.18 | 0.01 |
| 21 | 3.65 | 0.15 | 0.02 | 0.75 | 0.02 | 3.87 | 0.08 |
| 22 | 3.28 | 0.07 | 0.22 | 0.18 | 0.33 | 0.92 | 16.77 |
| 23 | 3.30 | 0.04 | 0.17 | 0.07 | 0.19 | 0.31 | 8.41 |
| 24 | 3.39 | 0.11 | 0.18 | 0.36 | 0.22 | 1.60 | 9.95 |
The triplet energy (ET) is another key parameter, which determines the potential performance of the host molecules applications in the phosphorescent blue emitters. It is evident from the previous reports,8,18,20 that the triplet energy should be higher than that of the phosphorescent guest to prevent back-transfer from the guest to the host. As mentioned for a blue emitter, the ET of the host should be higher than ∼2.8 eV. The calculated adiabatic ET (ZPVE) values of the host molecules are collected in Table 1, along with that of blue emitter (FIrpic) and reference host molecule (mCP). The computed ET values are in good agreement with experiment (Fig. 2) which may also reflect suitability of the method employed. From the Table 1, we can clearly see that all the Ph-Cb1-based host molecules exhibit higher ET value (∼0.13–0.39 eV) than that of FIrpic. Hence, all the molecules are suitable for the employment as a host material for the blue PhOLEDs. The calculated ET values of the para and meta-substituted hosts are in the range 2.71–2.89 and 2.82–2.98 eV, respectively. In order to understand, how the position of substituents affect the ET values of the hosts, we have compared meta-substituted host molecules with para-substituted molecules. The difference between the ET values of hosts 1, 2, 3, 4, 5 and 6 with 7, 8, 9, 10, 11 and 12, is 0.0, 0.1, 0.27, 0.01, 0.19, and 0.15 eV, respectively. These results clearly indicate that meta-substituted host molecules have higher (except 7) ET values compared to para-substituted systems. Further, results show that the substitution at the meta-position effectively increases the ET values. This difference in the ET values between the para- and meta-substituted hosts can be understood by examining NTO.34,39–42 Results as obtained from the NTO analysis of optimized T1-state geometries of the Ph-Cb1 based host molecules are displayed in ESI (Fig. S8).† It is possible to note that for the hosts 3, 4, 5 and 6, the hole–electron pairs of the NTOs correspond to a localized transition within the subunits (Ph-Cbz, BFP, BTP and Ph-Cb1). Hence, the ET value is close to that of those units. In host 1, the hole–electron pair of the NTOs is localized on the core. In the case of host 2, hole–electron pair can be seen on the biphenyl moiety. Therefore, the ET value is close to that of Cb1 and biphenyl units. In the case of para-substituted hosts, hole–electron pairs of the NTOs are delocalized (except host 7 and 10) on the biphenyl unit, which indicate that the biphenyl unit is responsible for the triplet state of these molecules. Hence, the ET values are close to that of the biphenyl moiety. In hosts 7 and 10, the hole–electron pairs of the NTOs are localized on the core Cb1 unit and thus ET values are close to that of core unit. It is possible to note that the ET values of the hosts materials are strongly influenced by the position of substitution rather than the substituted groups.
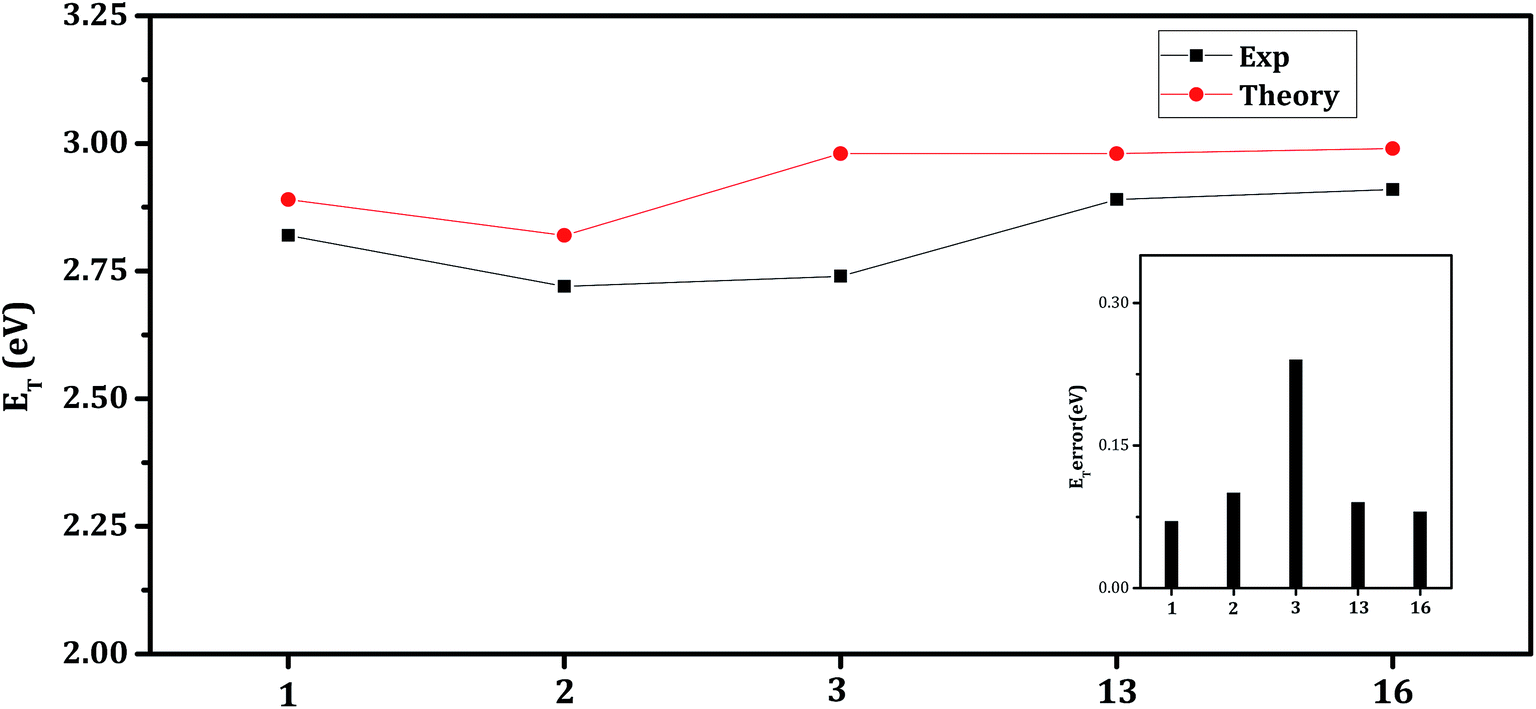 | ||
| Fig. 2 The calculated and experimental triplet energy (ET) of hosts 1, 2, 3, 13, and 16 along with the estimated error of predicted values in B3LYP/6-31G* method. | ||
The energy difference between the singlet and triplet energy states (ΔEST) is often used as a parameter to gauge the performance of the hosts, which is related to the triplet exciton formation and associated driving voltage.32,36,39,42 Generally, host molecule with lower ΔEST value is known to perform well. The calculated adiabatic ΔEST values of the para and meta-substituted host molecules are presented in the Table 1. The ΔEST values (0.49–0.77 eV) of all the designed hosts are lower than the reference host mCP (0.83 eV). Further, it can be noticed from the Table 1 that, the ΔEST values decrease (except hosts 11 and 12) when the subunits are substituted at the para-postion of the Ph-Cb1 core, when compared to the same group at the meta-position. Overall these findings indicate that triplet exciton formation is more effective in para-substituted hosts when compared to meta-position. The noteworthy point that emerge from the preceding discussion are as follows: (v) the para-substitution leads to better charge injections and small ΔEST values. (vi) The meta-substitution results in better charge transport and higher triplet energy.
3.2 Electronic structure of Ph-Cbz-based host molecules
As explained in Scheme 1, the subunits DBF, DBT, Ph-Cbz, DBF, BTP and Ph-Cb1 were substituted at the meta- (hosts 13, 14, 15, 16, 17, and 18) and para- (hosts 19, 20, 21, 22, 23, and 24) positions of the Ph-Cbz core leading to twelve host molecules. The calculated HOMO and LUMO values of the Ph-Cbz-based host molecules are displayed in Fig. 3 (also in ESI (Table S2†)) along with those of HTL, ETL and emissive layer. The HOMO energy levels of various Ph-Cbz based host molecules are destabilized (0.02–0.27 eV) when compared to their Ph-Cb1 based counterparts. This destabilization suggests that substitution at Ph-Cbz core may facilitate hole injection when compared to Ph-Cb1 based host molecules. The calculated HOMO (IP) energies for hosts 13–24, vary from −5.48 to −5.69 eV (6.41 to 6.99 eV), depending on the nature and position of the substitution. It can be observed from Fig. 3 that the HOMO energy levels of all the Ph-Cbz-based host molecules ranges −5.48 to −5.69 eV. Furthermore, these values are in between HTL (−4.91 eV) and emissive layer (−5.91 eV). The IP values (6.41 to 6.95 eV) of the Ph-Cbz-based hosts (expect host 13, 14, 16, 17) lie in between those of TAPC (5.80 eV) and FIrpic (6.96 eV). Hence, all these host molecules can efficiently transport the hole from HTL to the host and to the guest emitter. It is interesting to note that, all the para-substituted host materials (19–24) exhibit lower barrier for hole injection when compared with meta-substituted hosts. The IP values of all the host molecules (except 13, 14, 16, 17 and 22) are lower than the reference host materials (mCP, IP = 6.80 eV). This finding indicates that these hosts may have better hole transport and improved injection barrier for the hole than mCP. It can be seen from Fig. 3 that all the host molecules (except 13 and 21) have stabilized LUMO (0.01–0.9 eV) energy levels in contrast to Ph-Cb1-based hosts. The trend in the EA (0.03–0.08 eV) (Table 1) is also similar to that of LUMO energy levels. These values elicit that these hosts may have low injection barrier for the electron with respect to the Ph-Cb1-based hosts. Comparison of results from the calculations on the Ph-Cb1 and Ph-Cbz based host systems vividly reveals that the Ph-Cbz-based hosts may have better hole and electron injection properties.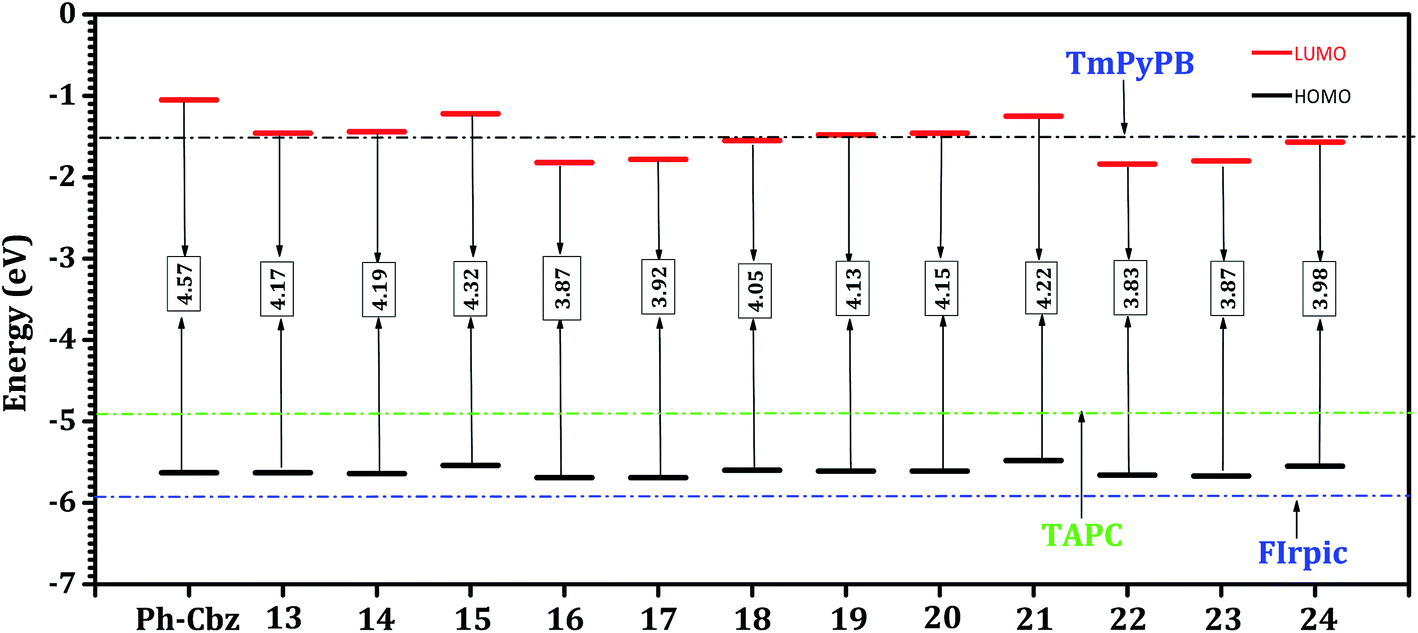 | ||
| Fig. 3 HOMO and LUMO energy levels (in eV) and HOMO–LUMO energy gap (Eg) of Ph-Cbz-based hosts and reference HTL (TAPC), ETL (TmPyPB), and emitter (FIrpic) as obtained at the B3LYP/6-31+G* level. | ||
To gain insight into the origin of these findings, we now focus on the HOMO and LUMO energy levels of the Ph-Cbz-based hosts and their core unit. A comparison of the HOMO–LUMO (H–L) gaps of Ph-Cbz and the designed hosts (Fig. 3) highlights that, the variations in the LUMO levels are more pronounced than in the corresponding HOMO levels in contrast to Ph-Cb1. The LUMO levels of all these systems are significantly stabilized by about ∼0.17–0.79 eV. Whereas, the range of destabilization of HOMO energies of these systems varies from ∼0.0–0.06 eV. The similar trends have been observed in both IP (0.04–0.62 eV) and EA (0.06–0.32 eV) values. These changes are also in accordance with the previous reports on the carbazole based hosts.40 Unlike Ph-Cb1-based hosts, the electron density distributions of HOMO (Fig. S9†) levels of all the Ph-Cbz-based hosts are localized on the Ph-Cbz core unit. Hence the HOMO energy is close to that of Ph-Cbz core. The LUMO levels are strongly stabilized owing to the localization of electron density on the subunits. The high stability of LUMO levels of 16, 17, 22 and 23 can be attributed to the localization of its LUMO on the BFP and BTP subunits. To gain further insights, a systematic comparison of HOMO (IP) and LUMO (EA) energy levels of hosts 13, 14, 15, 16, 17, and 18 with 19, 20, 21, 22, 23, and 24 has been undertaken. The HOMO levels of para-substituted hosts are destabilized by ∼0.02–0.06 eV energy levels unlike meta-substituted systems. The corresponding destabilization in the IP is ∼0.15–0.19 eV. Similar to Ph-Cb1-based hosts, the LUMO energy levels of Ph-Cbz systems are less sensitive. The maximum variation of LUMO (EA) energies between para and meta-substituted hosts ranges from 0.02–0.03 eV (0.02–0.05 eV).
The frontier molecular orbital (FMO) distributions and composition of the Ph-Cbz-based hosts are displayed in ESI (Table S6 and Fig. S9).† Results clearly elicit that, all the Ph-Cbz-based host materials have a clear charge separated state with an exception of host 21. The HOMO levels of all the systems are mainly localized on the Ph-Cbz (85.61–99.45%) core, due to strong electron donating nature of the Ph-Cbz unit, The LUMO levels are concentrated on the subunits (84.32–94.24%), due to their electron accepting nature. In case of host 21, HOMO is delocalized on the entire molecule and LUMO is delocalized on the phenyl and subunit. The calculated reorganization energies for hole (λh) and electron (λe) transport of the Ph-Cbz-based host molecules are listed in Table 1. For all the meta-substituted hosts (13–18), the reorganization energies for electron transport (λe = 0.17–0.37 eV) are marginally higher than those for hole transport (λh = 0.09–0.12 eV). These findings reveal that hole transport performance of these hosts is more favorable than the electron transport. This characteristics of hosts molecules are reinforced by the electron density distributions of MOs (Fig. 5). In the case of para-substituted hosts (13–24, except host 20), all the molecules have slightly higher λh (0.21–0.25 eV) values than those of λe (0.18–0.23 eV) implying ambipolar nature. For the hosts 14, 15 and 20, Δλ appreciably differs due to considerably higher λe values. These findings reiterate that linking topology influences both hole and electron reorganization energies. The calculated ranges in the hole mobilities for para and meta-substituted hosts are 0.29–11.58 and 0.03–4.18 cm2 V−1 s−1, respectively. The calculated electron mobilities for meta and para-substituted vary from 0.03–11.76 and 0.01–16.77 cm2 V−1 s−1, respectively.
The triplet energies (Table 1) of the all the host molecules have higher than that of FIrpic, by about ∼0.11–0.40 eV. Moreover, ET values of Ph-Cbz-based host molecules are almost the same as that of their Ph-Cb1 counterparts. However, 19, 21 and 22 are different from those of corresponding Ph-Cb1 counterparts. Similar to Ph-Cb1-based hosts, meta-substituted host molecules have higher ET values when compared to para-substituted systems due to the effective extended conjugation. These findings are also supported by the NTOs analysis on these systems (Fig. S10†). The calculated ΔEST values (Table 1) of the Ph-Cbz-based hosts materials range from 0.48–0.76 eV. Interestingly, all (except host 15) the meta-substituted hosts have lower ΔEST unlike the para-substituted molecules, which indicate that triplet exciton formation is more effective in meta-substituted hosts with reference to para-substituted systems. It is worth pointing out that the hosts 16 and 17 exhibit ΔEST as low as 0.48 eV. The noteworthy points that emerge from the preceding discussion are as follows: (vii) substitution at the para-position yields better charge injections and (viii) the same at the meta-position results in better charge transport, higher triplet energy and lower ΔEST values in Ph-Cbz based host molecules.
It is interesting to make inter-comparison between various host systems designed based on Ph-Cb1 and Ph-Cbz. It is possible to note that all the systems based on Ph-Cb1 do not have ambipolar property and associated balanced charge transport. The exceptions are 3, 4 and 5. However, these systems have more injection barrier for charge carriers. On the other hand, the hosts based on Ph-Cbz possess ambipolar (smaller HOMO–LUMO energy gap and clear charge separation) and balanced charge transport properties. As a consequence, these host molecules are potential candidates for blue PhOLEDs. Among the twenty four host molecules, hosts 16, 17, 18, 19, 22, 23, and 24 may outperform the other hosts due to their lower charge injection barrier for hole and electron, balanced charge transport and clear charge-separated state. However, these host materials have higher ΔEST value (except 16 and 17) than experimentally reported value for host 3.
It is evident from the previous study42 that further introduction of carbazole units (C–N linkage) at the 3, 6 positions of the Ph-Cbz core unit substantially decreases the ΔEST value. Moreover, in our previous work39 we have also systematically increased carbazole moiety at the carbazole core unit while fixing the carboline at the para-position to analyze change in the ΔEST values. Results revealed that di-substituted carbazole hosts exhibit lower ΔEST value when compared with mono-substituted carbazole moiety. The experimental results also reinforce the same findings.9 Hence, in order to reduce the ΔEST values of above-mentioned hosts, we have introduced the two carbazole units into the selected hosts (16, 17, 18, 19, 22, 23, and 24) and the corresponding molecules are shown in Scheme 2. In order to compare the results with the experimentally reported host, two carbazole units were substituted in the host 3 at 3, 6 positions. Findings from the calculations are presented in the following section.
 | ||
| Scheme 2 Chemical structure of newly designed host molecules. | ||
4. Potential host molecules for blue PhOLEDs
The hosts 25, 26, 27, 28, 29, 30, 31 and 32 were designed by introducing the carbazole (C–N linkage) units at the 3, 6 positions of the hosts 3, 16, 17, 18, 19, 22, 23, and 24, respectively. The array of electronic properties clearly reveal that the addition of the two carbazole units yields hosts (25, 26, 27, 28, 29, 30, 31 and 32) with improved electronic properties. Both HOMO and LUMO levels of these hosts (Fig. 4) indicate that these systems have improved injection barriers for hole and electron with reference to starting systems (3, 16, 17, 18, 19, 22, 23, and 24). It can be found that the addition of the two carbazole units destabilizes the HOMO levels (0.07 to 0.15 eV) and stabilizes the LUMO levels (0.12 to 0.48 eV) of these hosts. In addition, these energy levels are in alignment with the neighboring HTL, ETL and FIrpic. The comparison of IP and EA values provides similar findings (Table 3). The calculated IP values (6.32–6.38 eV) of the new host materials lie in between those of TAPC (IP = 5.80 eV) and FIrpic (IP = 6.96 eV). Hence, these hosts can potentially transfer holes from HTL to the host and to the guest emitter. The calculated EA values (Table 3) for the isolated host materials are found to be in the range of 0.70–0.89 eV, which are close to that of TmPyPB (EA = 0.68 eV).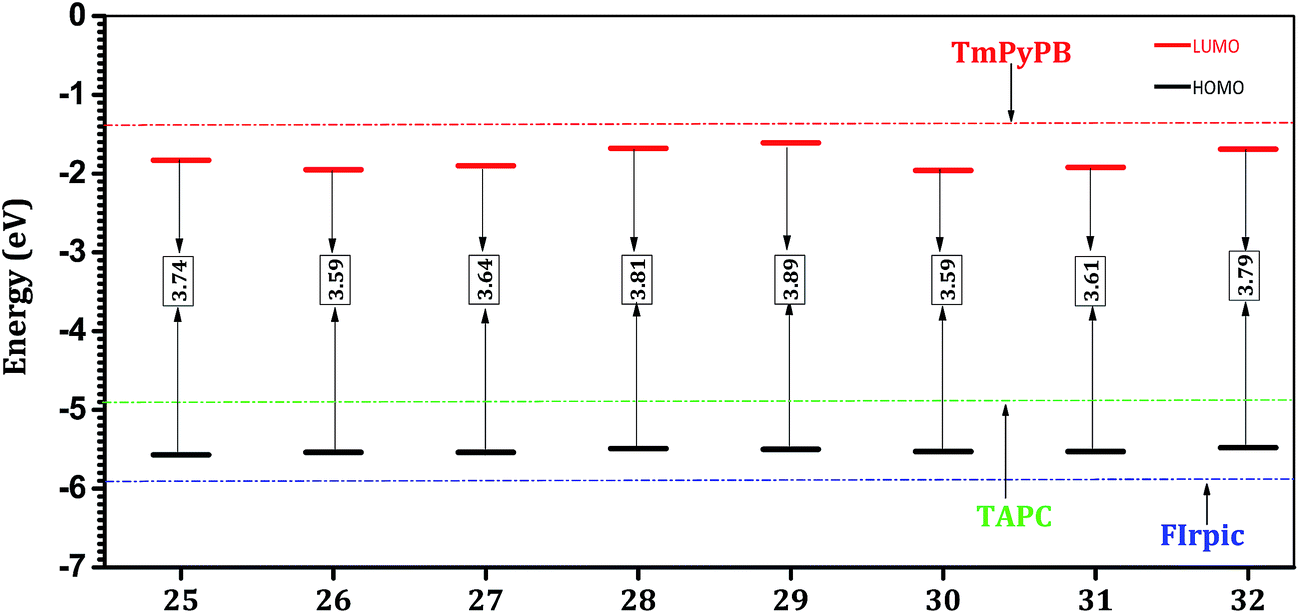 | ||
| Fig. 4 HOMO and LUMO energy levels (in eV) and HOMO–LUMO energy gap (Eg) of designed host materials with two carbazole units and reference HTL (TAPC), ETL (TmPyPB), and emitter (FIrpic) as obtained at the B3LYP/6-31+G* level. | ||
| Hosts | IP | EA | λh | λe | Δλ | ETa | E0Ta | ES | ΔEST |
|---|---|---|---|---|---|---|---|---|---|
| a ET and E0T denote the adiabatic triplet energy without and with zero-point vibrational (ZPVE) energy correction. | |||||||||
| 25 | 6.36 | 0.79 | 0.13 | 0.23 | 0.11 | 2.83 | 2.71 | 3.03 | 0.32 |
| 26 | 6.38 | 0.86 | 0.22 | 0.14 | 0.08 | 2.99 | 2.84 | 3.11 | 0.27 |
| 27 | 6.38 | 0.85 | 0.22 | 0.15 | 0.07 | 3.09 | 2.92 | 3.25 | 0.33 |
| 28 | 6.32 | 0.73 | 0.23 | 0.15 | 0.08 | 2.98 | 2.84 | 3.26 | 0.43 |
| 29 | 6.33 | 0.70 | 0.22 | 0.15 | 0.07 | 2.98 | 2.83 | 3.25 | 0.42 |
| 30 | 6.37 | 0.89 | 0.22 | 0.14 | 0.08 | 2.98 | 2.83 | 3.24 | 0.41 |
| 31 | 6.37 | 0.88 | 0.21 | 0.15 | 0.06 | 2.82 | 2.71 | 3.14 | 0.43 |
| 32 | 6.30 | 0.76 | 0.21 | 0.15 | 0.06 | 2.83 | 2.71 | 3.32 | 0.61 |
It can be seen from Fig. 5 that all the host materials with two Cbz units exhibit a clear charge-separated state. The calculated ET values of these hosts are presented in Table 3 which reveal that these hosts are nearly identical to their corresponding Ph-Cbz based hosts (except 25). The lower ET value of the host 25 is attributed to the localization of hole–electron wave function on the Cb1 unit (Fig. 6). All the host materials have higher ET values than that of reference emitter FIrpic (2.59 eV). The reorganization energies for hole are slightly higher than those of electron (Table 3) which implies that the electron transport is favorable. The difference in the reorganization energies of the hole and electron ranges from 0.06–011 eV. Thus, it is possible to note that the addition of Cbz units has negligible impact on the charge transport properties. The calculated hole and electron mobilities (Table 4) of these host molecules are found to be 0.72–14.80 and 0.06–10.90 cm2 V−1 s−1, respectively.
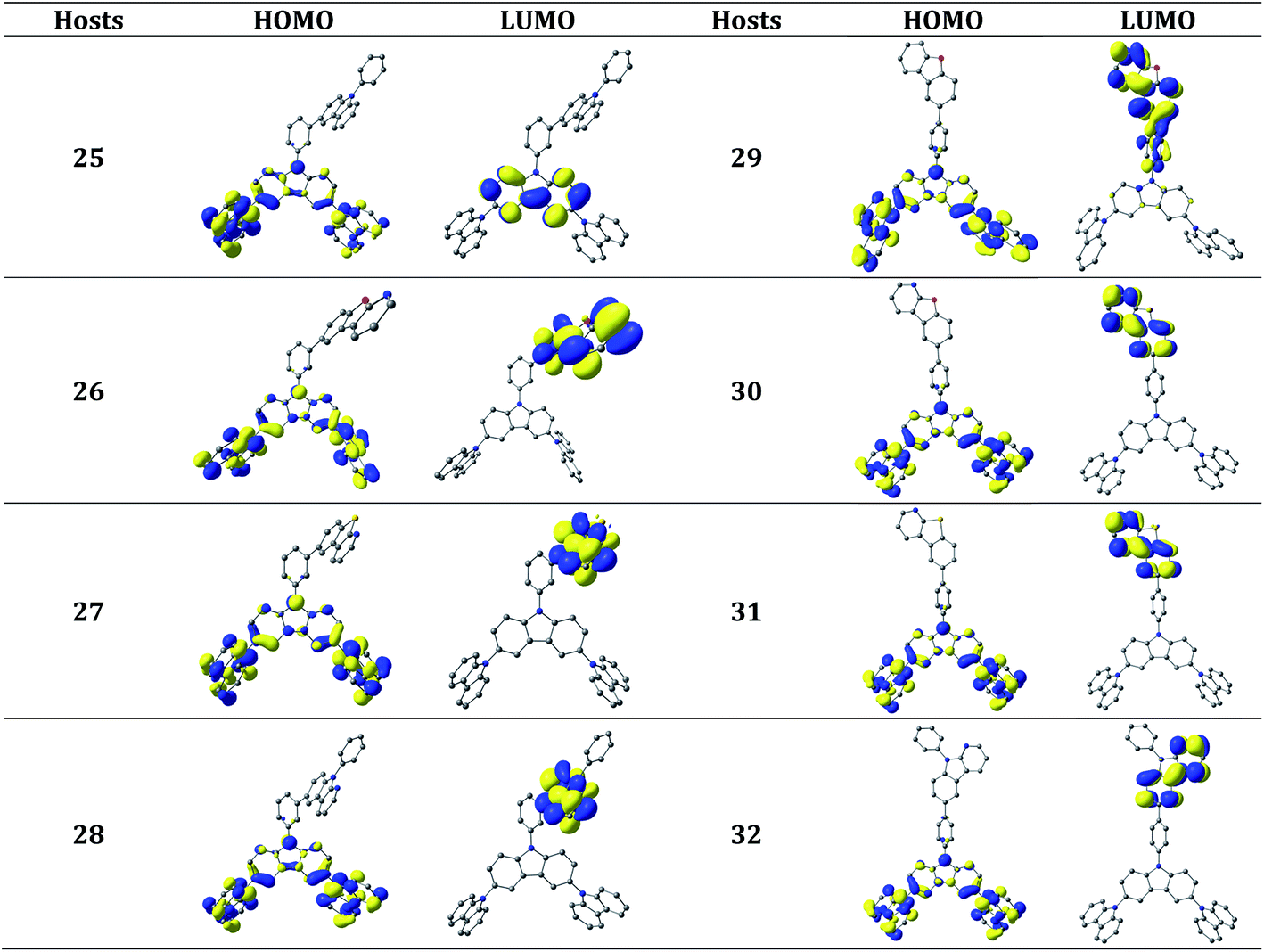 | ||
| Fig. 5 The contour plots (isosurface value = 0.03 au) of the HOMO and LUMO levels of the new host molecules with two carbazole unit. The hydrogen atoms are omitted here for clarity. | ||
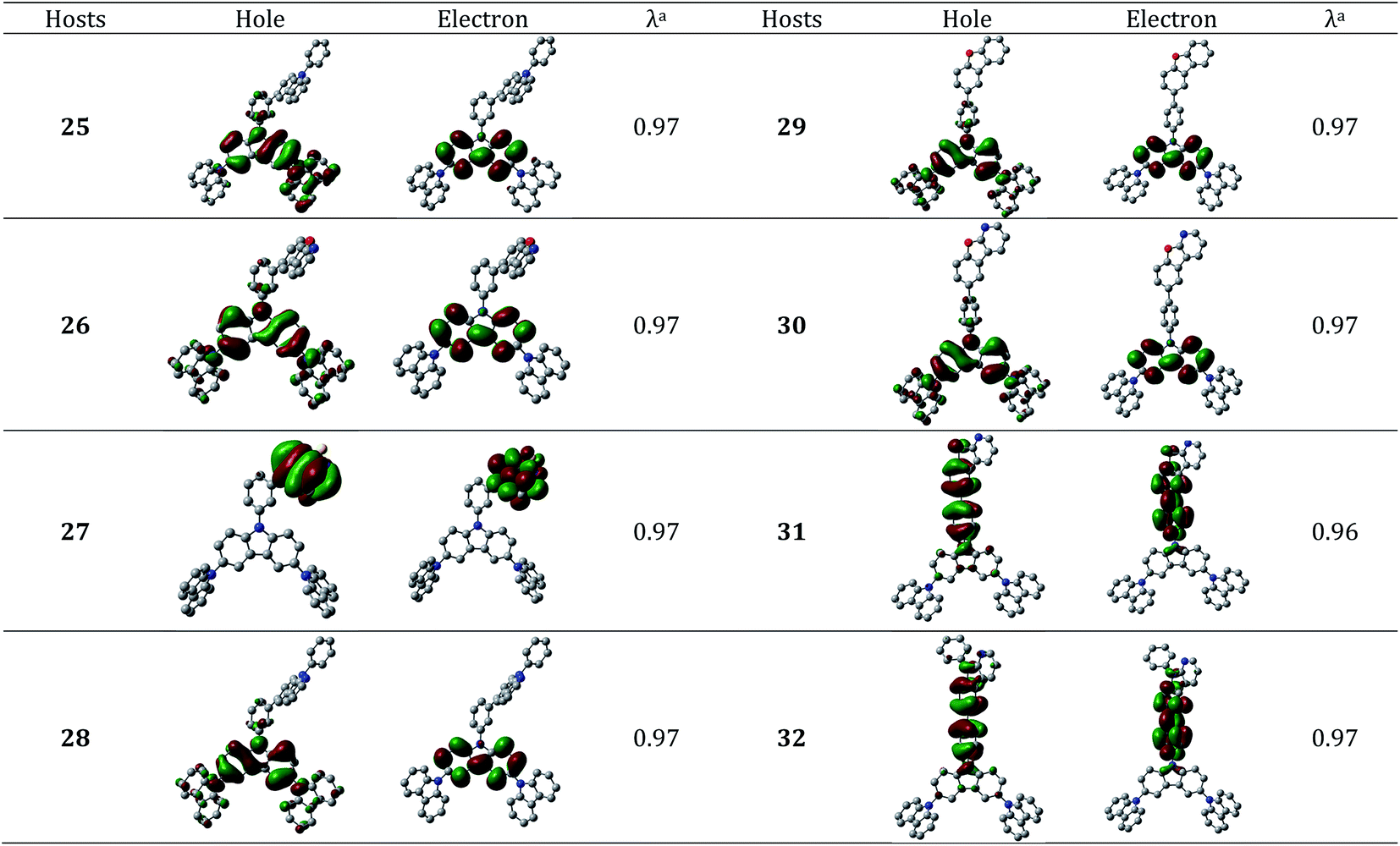 | ||
| Fig. 6 Hole–particle NTOs (isosurface value = 0.030 au) of the new host molecules with two carbazole unit. The hydrogen atoms are omitted here for clarity. aλ represents natural transition orbital eigenvalue. | ||
| Hosts | d (Å) | t (eV) | K (s−1) × 10−14 | μ (cm2 V−1 s−1) | |||
|---|---|---|---|---|---|---|---|
| Hole | Electron | Hole | Electron | Hole | Electron | ||
| 25 | 3.54 | 0.09 | 0.02 | 1.06 | 0.01 | 5.20 | 0.07 |
| 26 | 3.58 | 0.08 | 0.01 | 0.27 | 0.01 | 1.34 | 0.06 |
| 27 | 3.40 | 0.19 | 0.04 | 1.52 | 0.16 | 6.84 | 0.73 |
| 28 | 3.72 | 0.06 | 0.03 | 0.13 | 0.09 | 0.72 | 0.49 |
| 29 | 3.33 | 0.24 | 0.13 | 2.42 | 1.70 | 10.46 | 7.34 |
| 30 | 3.29 | 0.11 | 0.15 | 0.50 | 2.58 | 2.15 | 10.90 |
| 31 | 3.58 | 0.25 | 0.08 | 2.97 | 0.64 | 14.80 | 3.22 |
| 32 | 3.49 | 0.25 | 0.28 | 2.97 | 7.89 | 14.07 | 3.74 |
The calculated adiabatic ΔEST values of these host molecules are presented in Table 3. It is noteworthy to mention that all the host molecules with two Cbz units have lower ΔEST (0.27 to 0.61 eV) values than that of the Ph-Cbz based hosts (0.48 to 0.69 eV). It is clearly evident from these results that the introduction of two Cbz units at the 3, 6 positions of the parent hosts substantially reduces the difference in the singlet–triplet energies. This variation in the ΔEST values is due to the profound changes in the adiabatic singlet energy levels (ES) when compared to the adiabatic triplet energies (E0T). The variations in the ES and E0T energies range from 0.04–0.49 eV and 0.00–0.32 eV respectively. The substantial changes in the singlet energy due to singlet excited states are found to be dominated by a HOMO → LUMO transition with strong intramolecular charge transfer (ICT) character, i.e., from a HOMO localized on the newly introduced carbazoles to a LUMO localized on the subunits (Fig. 5). A similar observation is reported in the literature.42 The incorporation of two additional Cbz units is found to stabilize the singlet excited state leading to decrease in the ΔEST value. Overall, the hosts 25, 26, 27, 28, 29, 30, 31 and 32 stand out as the most efficient molecules based on the consideration of necessary electronic properties for the development of host molecules.
5. Conclusions
We have carried out a comprehensive DFT study on the structure and electronic properties of a series of carbazole and carboline based ambipolar host molecules. Thirty two host molecules have been designed by taking phenyl carbazole and phenyl α-carboline as the core units and dibenzofuran, dibenzothiophene, phenylcarbazole, benzofuropyridine, benzothiopyridine and phenyl pyridoindole as subunits. The density functional theory calculations were employed to screen the designed hosts based on an array of electronic properties, viz., ET, HOMO and LUMO, ΔEST, charge injection barriers, and charge mobility from the reorganization energies and charge transfer integral. The salient findings from this investigation are summarized in Fig. 7. The following important points emerge from this study: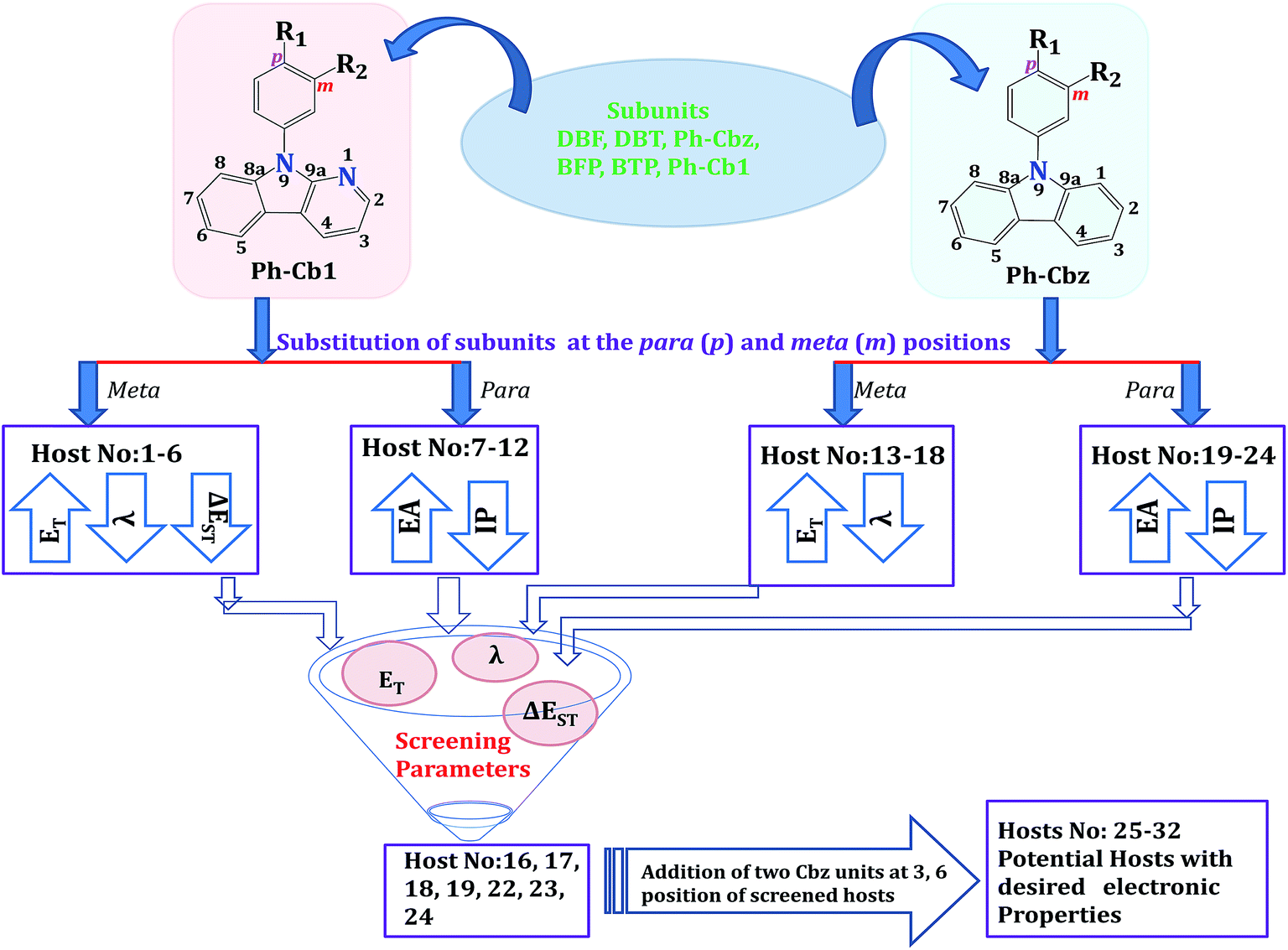 | ||
| Fig. 7 Flow chart of screened host materials for blue PhOLEDs. | ||
(1) The substitution at the para-position of Ph-Cb1 core leads to host molecules with good charge injection characteristics and small ΔEST values. On the other hand, the substitution at m-position yields better charge transport properties and higher triplet energy.
(2) The substitution of the subunits at the para-position of Ph-Cbz core enhances charge injection.
(3) The substitution at meta-position facilitates good charge transport properties, lower ΔEST and higher triplet energy.
(4) Overall, the substitution of subunits at the Ph-Cbz core yields host molecules with improved electronic properties when compared to the same units at Ph-Cb1 core.
(5) The main drawback of the Ph-Cbz based host molecules is that these hosts display higher ΔEST values than that of experimentally reported for host 3. This drawback can be overcome by incorporation of two additional carbazole units at the 3, 6 positions of the host molecules. The resultant host molecules exhibit lower singlet–triplet energy differences.
(6) Amongst, the newly designed hosts, molecules 25, 26, 27, 28, 29, 30, 31 and 32 are found to be promising hosts with lower barrier for hole and electron injection, balanced charge transport, the presence of clear separation of HOMO and LUMO (charge-separated state), and lower ΔEST values when compared to the experimentally reported value for host 3. Thus, the present computational investigation provides several promising lead molecules for the future development of new class of carbazole and carboline based host molecules for blue PhOLEDs.
Acknowledgements
This work was supported by the Technologies and Products for Solar Energy Utilization through Networks (CSIR-TAPSUN, NWP-55) programme. E. V. thanks AcSIR for enrollment in the Ph.D program. D. V. thanks DST, New Delhi, for financial support in the form of INSPIRE Faculty Award.References
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS.
- M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10967 CrossRef CAS.
- R. J. Holmes, S. R. Forrest, Y. J. Tung, R. C. Kwong, J. J. Brown, S. Garon and M. E. Thompson, Appl. Phys. Lett., 2003, 82, 2422 CrossRef CAS.
- S. Tokito, T. Iijima, Y. Suzuri, H. Kita, T. Tsuzuki and F. Sato, Appl. Phys. Lett., 2003, 83, 569 CrossRef CAS.
- D. Kim, S. Salman, V. Coropceanu, E. Salomon, A. B. Padmaperuma, L. S. Sapochak, A. Kahn and J.-L. Brédas, Chem. Mater., 2010, 22, 247 CrossRef CAS.
- L. S. Sapochak, A. B. Padmaperuma, X. Y. Cai, J. L. Male and P. E. Burrows, J. Phys. Chem. C, 2008, 112, 7989 CAS.
- M.-H. Tsai, Y.-H. Hong, C.-H. Chang, H.-C. Su, C.-C. Wu, A. Matoliukstyte, J. Simokaitiene, S. Grigalevicius, J. V. Grazulevicius and C.-P. Hsu, Adv. Mater., 2007, 19, 862 CrossRef CAS.
- K. Brunner, A. van Dijken, H. Börner, J. J. A. M. Bastiaansen, N. M. M. Kiggen and B. M. W. Langeveld, J. Am. Chem. Soc., 2004, 126, 6035 CrossRef CAS PubMed.
- A. van Dijken, J. J. A. M. Bastiaansen, N. M. M. Kiggen, B. M. W. Langeveld, C. Rothe, A. Monkman, I. Bach, P. Stössel and K. Brunner, J. Am. Chem. Soc., 2004, 126, 7718 CrossRef CAS PubMed.
- I. Avilov, P. Marsal, J. L. Bŕedas and D. Beljonne, Adv. Mater., 2004, 16, 1624 CrossRef CAS.
- P.-I. Shih, C.-L. Chiang, A. K. Dixit, C.-K. Chen, M.-C. Yuan, R.-Y. Lee, C.-T. Chen, E. W.-G. Diau and C.-F. Shu, Org. Lett., 2006, 8, 2799 CrossRef CAS PubMed.
- M.-H. Tsai, H.-W. Lin, H.-C. Su, T.-H. Ke, C.-C. Wu, F.-C. Fang, Y.-L. Liao, K.-T. Wong and C.-I. Wu, Adv. Mater., 2006, 18, 1216 CrossRef CAS.
- X. Cai, A. B. Padmaperuma, L. S. Sapochak, P. A. Vecchi and P. E. Burrows, Appl. Phys. Lett., 2008, 92, 083308 CrossRef.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169 CrossRef CAS PubMed.
- L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137 CrossRef CAS PubMed.
- A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876 CrossRef CAS PubMed.
- L. Ding, S.-C. Dong, Z.-Q. Jiang, H. Chen and L.-S. Liao, Adv. Funct. Mater., 2015, 25, 645 CrossRef CAS.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484 RSC.
- D. Kim, S. Salman, V. Coropceanu, E. Salomon, A. B. Padmaperuma, L. S. Sapochak, A. Kahn and J.-L. Brédas, Chem. Mater., 2010, 22, 247 CrossRef CAS.
- S. Gong, Q. Fu, W. Zeng, C. Zhong, C. Yang, D. Ma and J. Qin, Chem. Mater., 2012, 24, 3120 CrossRef CAS.
- S.-J. Su, H. Sasabe, H. Takeda and J. Kido, Chem. Mater., 2008, 20, 1691 CrossRef CAS.
- H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621 CrossRef CAS.
- D. R. Lee, C. W. Lee and J. Y. Lee, J. Mater. Chem. C, 2014, 2, 7256 RSC.
- B. Pan, H. Huang, X. Yang, J. Jin, S. Zhuang, G. Mu and L. Wang, J. Mater. Chem. C, 2014, 2, 7428 RSC.
- M. Kim, S. K. Jeon, S.-H. Hwanga and J. Y. Lee, Phys. Chem. Chem. Phys., 2015, 17, 13553 RSC.
- P. Kautny, D. Lumpi, Y. P. Wang, A. Tissot, J. Bintinger, E. Horkel, B. Stoger, C. Hametner, H. Hagemann, D. G. Ma and J. Frohlich, J. Mater. Chem. C, 2014, 2, 2069 RSC.
- L. S. Sapochak, A. B. Padmaperuma, P. A. Vecchi, H. Qiao and P. E. Burrows, Proc. SPIE, 2006, 57, 6333 Search PubMed.
- J. Yin, S. L. Zhang, R. F. Chen, Q. D. Ling and W. Huang, Phys. Chem. Chem. Phys., 2010, 12, 15448 RSC.
- X. Gu, H. Zhang, T. Fei, B. Yang, H. Xu, Y. Ma and X. Liu, J. Phys. Chem. A, 2010, 114, 965 CrossRef CAS PubMed.
- J. Wu, Y. Liao, S. Wu, H. Li and Z. Su, Phys. Chem. Chem. Phys., 2012, 14, 1685 RSC.
- P. Marsal, I. Avilov, D. A. da Silva Filho, J. L. Brédas and D. Beljonne, Chem. Phys. Lett., 2004, 392, 521 CrossRef CAS.
- J. Wu, Y.-H. Kan, Y. Wu and Z.-M. Su, J. Phys. Chem. C, 2013, 117, 8420 CAS.
- X. Zhang, W. Shen, H. Sun, P. Yu, R. He and M. Li, J. Phys. Org. Chem., 2015, 28, 554 CrossRef CAS.
- M.-K. Yan, Y. Tao, R.-F. Chen, C. Zheng, Z.-F. An and W. Huang, RSC Adv., 2012, 2, 7860 RSC.
- J. Li, Z. Nie, H. Li, Y. Peng, Z. Wang, Z. Maia and W. Zheng, J. Mater. Chem. C, 2015, 3, 4859 RSC.
- E. Varathan, D. Vijay, P. S. V. Kumar and V. Subramanian, J. Mater. Chem. C, 2013, 1, 4261 RSC.
- E. Varathan, D. Vijay and V. Subramanian, J. Phys. Chem. C, 2014, 118, 21741 CAS.
- D. Kim, L. Zhu and J.-L. Brédas, Chem. Mater., 2012, 24, 2604 CrossRef CAS.
- D. Kim, V. Coropceanu and J. Brédas, J. Am. Chem. Soc., 2011, 133, 17895 CrossRef CAS PubMed.
- S. Salman, D. Kim, V. Coropceanu and J.-L. Brédas, Chem. Mater., 2011, 23, 5223 CrossRef CAS.
- H. Fukagawa, N. Yokoyama, S. Irisa and S. Tokito, Adv. Mater., 2010, 22, 4775 CrossRef CAS PubMed.
- C. W. Lee and J. Y. Lee, Adv. Mater., 2013, 25, 5450 CrossRef CAS PubMed.
- C. W. Lee, Y. Im, J. A. Seo and J. Y. Lee, Chem. Commun., 2013, 49, 9860 RSC.
- C. W. Lee and J. Y. Lee, Chem. Mater., 2014, 26, 1616 CrossRef CAS.
- T. Motoyama, H. Sasabe, Y. Seino, J.-I. Takamatsu and J. Kido, Chem. Mater., 2014, 26, 1616 CrossRef.
- S. J. Kim, Y. J. Kim, Y. H. Son, J. A. Hur, H. A. Um, J. Shin, T. W. Lee, M. J. Cho, J. K. Kim, S. Joo, J. H. Yang, G. S. Chae, K. Choi, J. H. Kwon and D. H. Choi, Chem. Commun., 2013, 49, 6788 RSC.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4218 CrossRef CAS PubMed.
- Y. H. Son, Y. J. Kim, M. J. Park, H. Y. Oh, J. S. Park, J. H. Yang, M. C. Suh and J. H. Kwon, J. Mater. Chem. C, 2013, 1, 5008 RSC.
- C. W. Lee, Y. Im, J. A. Seo and Y. Lee, Chem. Commun., 2013, 49, 9860 RSC.
- C. W. Lee and J. Y. Lee, Chem. Commun., 2013, 49, 6185 RSC.
- Y. Im and J. Y. Lee, Chem. Commun., 2013, 49, 5948 RSC.
- C. W. Lee and J. Y. Lee, Chem. Commun., 2013, 49, 1446 RSC.
- C. W. Lee and J. Y. Lee, Adv. Mater., 2013, 25, 596 CrossRef CAS PubMed.
- C. W. Lee, J. A. Seo, M. S. Gong and J. Y. Lee, Chem.–Eur. J., 2013, 19, 1194 CrossRef CAS PubMed.
- C. W. Lee and J. Y. Lee, Dyes Pigm., 2014, 109, 1 CrossRef CAS.
- C. W. Lee and J. Y. Lee, Dyes Pigm., 2014, 103, 34 CrossRef CAS.
- C. W. Lee, J.-K. Kim, S. H. Joo and J. Y. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 2169 CAS.
- C. W. Lee, Y. Im, J. A. Seo and J. Y. Lee, Org. Electron., 2013, 14, 2687 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- B. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef.
- D. Jacquemin, E. A. Perpète, I. Ciofini and C. J. Adamo, J. Chem. Theory Comput., 2010, 6, 1532 CrossRef CAS PubMed.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420 CrossRef CAS PubMed.
- T. Ziegler, A. Rauk and E. J. Baerends, Theor. Chim. Acta, 1977, 43, 261 CrossRef CAS.
- T. Kowalczyk, S. R. Yost and T. V. Voorhis, J. Chem. Phys., 2011, 134, 054128 CrossRef PubMed.
- N. A. Besley, A. T. B. Gilbert and P. M. W. Gill, J. Chem. Phys., 2009, 130, 124308 CrossRef PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- B. C. Lin, C. P. Cheng, Z. Q. You and C. P. Hsu, J. Am. Chem. Soc., 2005, 127, 66 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. Da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926 CrossRef CAS PubMed.
- J. L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971 CrossRef PubMed.
- R. A. Marcus, J. Chem. Phys., 1965, 43, 679 CrossRef CAS.
- R. A. Macus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265 CrossRef.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Phys., 2005, 123, 161103 CrossRef PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- R. M. Kumar, M. Elango and V. Subramanian, J. Phys. Chem. A, 2010, 114, 4313 CrossRef CAS PubMed.
- T. Koopmans, Physica, 1934, 1, 104 CrossRef.
- H. Sahu and A. N. Panda, Phys. Chem. Chem. Phys., 2014, 16, 8563 RSC.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 167, 12866 Search PubMed.
- E. F. Valeev, V. Coropceanu, D. A. da Silva Filho, S. Salman and J. L. Brédas, J. Am. Chem. Soc., 2006, 128, 9882 CrossRef CAS PubMed.
- J.-L. Brédas, Mater. Horiz., 2014, 1, 17 RSC.
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M.-a. Kakimoto, Adv. Funct. Mater., 2008, 18, 584 CrossRef CAS.
- The calculated mobility value is based on very simple approximation (dimer splitting method). Moreover, we have not considered the orbital orthogonalization and site energy correction. As a result, the calculated mobility values are expected to be somewhat overestimated when compared to experimental value. Hence, caution should be taken when draw a conclusion from these calculated values.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15748c |
| This journal is © The Royal Society of Chemistry 2016 |