
Reactivity of a half-lantern Pt2(II,II) complex with triphenylphosphine: selectivity in a protonation reaction†
Maryam Niazia,
Hamid R. Shahsavari *a,
Mohsen Golbon Haghighib,
Mohammad Reza Halvagarc,
Samaneh Hatamia and
Behrouz Notashb
*a,
Mohsen Golbon Haghighib,
Mohammad Reza Halvagarc,
Samaneh Hatamia and
Behrouz Notashb
aDepartment of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), Yousef Sobouti Blvd., Zanjan 45137-6731, Iran. E-mail: shahsavari@iasbs.ac.ir
bDepartment of Chemistry, Shahid Beheshti University, Evin, Tehran 19839-69411, Iran
cChemistry & Chemical Engineering Research Center of Iran, Tehran, 14968-13151, Iran
First published on 8th August 2016
Abstract
The half-lantern Pt2(II,II) complex [{Pt(ppy)(μ2-Spy)}2], 1, in which ppyH = 2-phenylpyridine and pySH = pyridine-2-thiol, is treated with two equivalents of triphenylphosphine ligand (dissociated Pt–NSpy bonds) to form complex [Pt(ppy)(η1-S-Spy)(PPh3)], 2. This complex can alternatively be obtained directly from the reaction of complex [Pt(ppy)(PPh3)Cl], 3, with an ethanolic solution of sodium pyridine-2-thiolate. The transphobia effect (T) controls the geometry of complex 2 and spectroscopic data confirms that the phosphine ligand is positioned trans to the nitrogen atom of the cyclometalating fragment (ppy). The solution dynamics of complex 2 was investigated by NMR spectroscopy and it revealed a fluxional behavior in solution. Besides, the protonation reaction conditions for complex 2 revealed the sensitivity of this complex to the anionic group of the acidic species. For instance, reaction of complex 2 with hydrogen chloride that contains a coordinating anion, resulted in a platinum–sulfur bond cleavage and subsequent formation of the corresponding chloride complex 3. In contrast, upon treatment of complex 2 with HPF6, involving a non-coordinating counter-anion, the pendant nitrogen atom was protonated and complex [Pt(ppy)(η1-S-Spy*) (PPh3)][PF6], 5, was obtained.
Introduction
The divalent platinum(II) complexes of the general formula, [{Pt(C^N) (μ2-SN)}2], [C^N = different cyclometalating units and SN = various 2-thiolate ligands], are known as neutral half-lantern complexes.1 Generally, these type of complexes display an intense 3MMLCT (Metal–Metal to Ligand Charge Transfer) emission2–5 and this property could play an important role in their applications. For instance, they are used as phosphorescent dopants for the preparation of OLEDs,6–9 and they are good candidates for detection of poisonous ions10 such as Hg2+ or they are used in oxidative addition reactions.11 In the later example, half-lantern complexes undergo two-electron oxidation upon reaction with high or medium polar species to give the corresponding Pt(III)–Pt(III) complexes.1–3,7,8,11–15 The results of this type of oxidation reaction is known to be non-emissive.16In spite of different reactivity of such half-lantern platinum(II) complexes in oxidative–addition reaction,11 these complexes are potential candidates in other important reactions, for example in ligand substitution reactions. These complexes contain weak platinum–nitrogen bonds3 and upon treatment with strong donor ligands these weak bonds break up17,18 and may lead to the formation of interesting compounds that encompass a heterocyclic thiolate group with free nitrogen atom. However, both nitrogen and sulfur donor atoms in these complexes can act as coordinating sites.19–21 The coordination chemistry of such complexes can be complicated19,20 and the main attractive point for preparation of these compounds is potential application in diverse area such as construction of luminescent materials,22,23 catalysis,24,25 and biology.26,27
We explored the reactivity of complex [{Pt(ppy)(μ2-Spy)}2], 1, ppyH = 2-phenylpyridine and pySH = pyridine-2-thiolate, with PPh3 and complex [Pt(ppy)(η1-S-Spy) (PPh3)], 2, was obtained in acetone solution. The geometry of complex 2 was estimated according to spectroscopic methods and the transphobia (T) effect,28–31 which is associated with the trans influence of the σ platinum–carbon bond existent in this complex. We found out that complex 2 displayed anionic selectivity upon its protonation reaction with acidic species. When this complex was treated with HCl, the Pt–S bond of thiolate ligand was cleaved. In contrast, when complex 2 was reacted with HPF6, the nitrogen atom of pyridyl group was protonated.
Result and discussion
Reactivity of complex 1 with triphenylphosphine: synthesis and characterization of complex 2
The molecular structure3,32 of binuclear half-lantern Pt2(II,II) complex [{Pt(ppy)(μ2-Spy)}2], 1, reveals the geometry of each Pt(ppy) moiety that are linked by two pyridine-2-thiolate groups in a head to tail fashion. The high trans influence of Pt–C bonds resulted in an anti conformation and the nitrogen atom of Spy ligand occupies trans position of the carbon atom.2,3 Therefore, the Pt–NSpy distances in this configuration are considerably long (∼2.14 Å)3,32 and these bonds easily dissociate with strong nucleophiles such as phosphorus donor ligands.As shown in Scheme 1, the reaction of complex 1 with 2 equivalents of PPh3, proceeded by dissociation of the nitrogen atoms33–35 of Spy bridging ligands, resulting in complex [Pt(ppy) (η1-S-Spy)(PPh3)], 2, in high yield.
 | ||
| Scheme 1 Reaction of complex 1 with phosphine ligand in different dry solvents and under Ar atmosphere. | ||
The substitution reaction described in Scheme 1 only proceeded in dry solvent and under inert argon atmosphere. This is because of the phosphine ligand being sensitive to water content in the solvent and molecular oxygen in the environment.36–38 The type of solvent also plays a major role in the overall outcome of the reaction. When non-halogenated solvents like acetone or dimethylsulfoxide (DMSO) were used, PPh3 ligand clearly ruptured Pt–NSpy bonds in complex 1 and produced complex 2. Phosphine displacement did not occur if halogenated solvents such as chloroform (CHCl3) or dichloromethane (CH2Cl2) was used as reaction media. Therefore, reaction of complex 1 with halogenated solvents yielded corresponding Pt2(III,III)X2 complex [{Pt(ppy)(μ2-Spy)Cl}2], 1a (Scheme 1)2,3,11 and phosphine ligand did not play any role in this reaction. It is notable that oxidation reaction leading to complex 1a could happen in the absence of PPh3.8,11 Therefore, the parallel oxidation reaction competes with desired bond breaking reaction. This observation suggested that complex 1 require a vacant site for initial coordination of PPh3 ligand to the platinum center.17,18,39 To confirm this hypothesis, the substitution reaction between complex 1a, which lacks an open coordination site, and the phosphine ligand was investigated. This reaction did not yield any product and only PPh3 ligand was converted to phosphine oxide,36 (Fig. S1 and S2†).
Further investigation was carried out using UV-vis spectroscopy. In this study, complex 1 displayed a distinctive band centered at 500 nm (acetone), which was assigned to 1MMLCT transition and believed to be the main reason for the red color of complex 1 (Fig. 1).3 This band was used to monitor the substitution reaction. After addition of an excess amount of phosphine ligand to a solution of complex 1, the intensity of the peak decreased progressively, finally vanished and at the same time a new band was observed at 420 nm (Fig. 1 and S3†). According to time-dependent DFT (TD-DFT) calculations (see below), and previous assignments this new profile can be attributed to an admixture of 1L'LCT/1MLCT (Ligand (Spy) to Ligand (ppy) Charge Transfer/Metal to Ligand (ppy) Charge Transfer) transitions.23,40 Besides, the red color of mixture gradually disappeared and eventually a yellow solution was obtained. Therefore, the new band at 420 nm and the appearance of yellow color confirmed the formation of complex 2 and complete consumption of complex 1 (Fig. 1 and S3†).
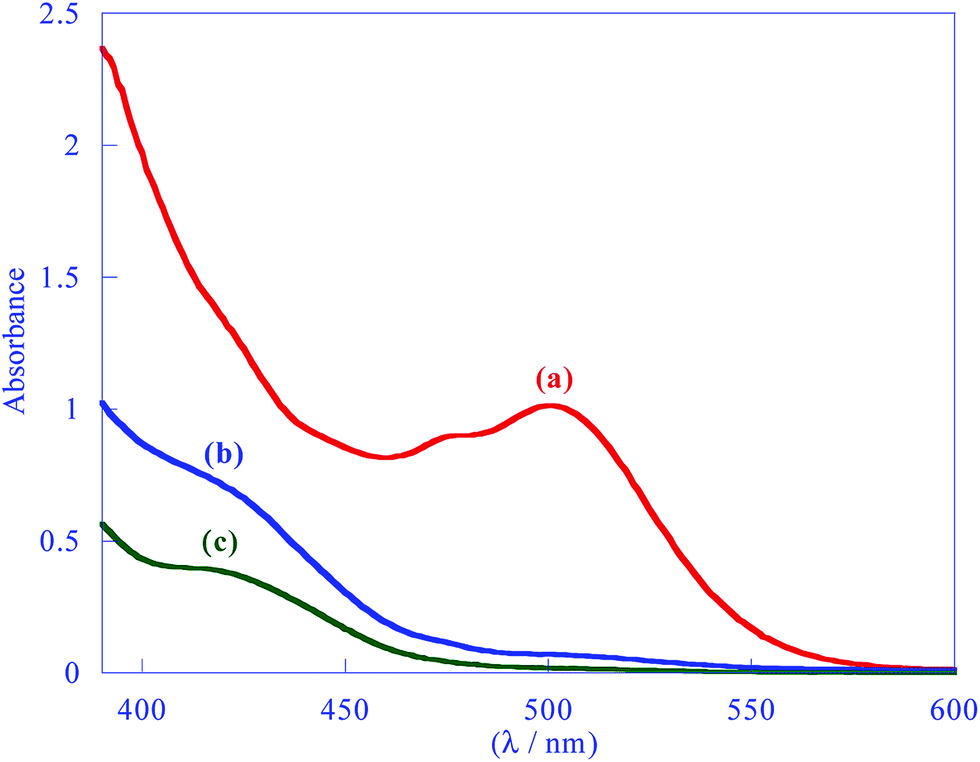 | ||
| Fig. 1 UV–vis spectra of (a) complex [{Pt(ppy)(μ2-Spy)}2], 1, (3 × 10−4 M); (b) the end point of reaction of excess PPh3 with complex [{Pt(ppy)(μ2-Spy)}2], 1, (c) the synthesized complex [Pt(ppy)(η1-S-Spy)(PPh3)], 2, (2.5 × 10−4 M) in acetone at 25 °C. | ||
To deduce the nature of the lower energy electronic transition of complex 2, TD-DFT calculations were conducted in acetone solution. The frontier molecular orbitals involved in this transition were shown in Fig. S4.† The relative compositions of these energy levels in terms of composing fragments are collected in Table S1.† In complex 2, the HOMO is mainly localized on the pyridine-2-thiolate (80%) ligand with partial contribution of the platinum (12%) metal center. The LUMO is predominantly located on the ppy (86%) cyclometalating ligand (Fig. S4 and Table S1†). The calculated value for the low energy absorption 433 nm is close to the experimental value of 420 nm, with a small red shift. The main contribution to this low energy profile (∼70%) including the HOMO → LUMO transition (Table S2†), can be allocated to a mixture of 1MLCT (Pt → ppy) with a notable contribution of 1L'LCT (Spy → ppy).
Complex 2 was also obtained using a different method via treatment of complex [Pt(ppy)(PPh3)Cl], 3,41,42 with an ethanolic solution of sodium pyridine-2-thiolate (NaC5H4NS) under Ar atmosphere (Scheme 2, step b').
 | ||
| Scheme 2 Potential routes for the preparation of complex 2 using complex A as a precursor. | ||
The above two reaction routes demonstrate an interesting point. As presented in Scheme 2, complexes 132 (step a) and 341,42 (step a') were prepared by similar platinum(II) starting complex [Pt(ppy)(DMSO)(Cl)], A.43 This suggests that in the first step, addition of either phosphine or sodium thiolate ligand to complex A was not crucial. Consequently, path (I) and (II) yielded the same complex 2 at the end of reactions (Scheme 2).
Complex 2 was inferred from common analytical methods (Elemental analyses and Mass) and its integrity in solution was studied by multinuclear (1H, 31P {1H}, in different solvents) NMR spectroscopy. The details are given in the Experimental section.
The ESI-MS spectrum of complex 2 (positive ions), illustrated three main fragment peaks assigned to the Pt species [Pt(ppy)(η1-S-SpyH)(PPh3)]+ (m/z 722, 100%), [Pt(ppy)(PPh3)]+ (m/z 611, 94%) and [{Pt(ppy)(PPh3)}2(μ2-Spy)]+ (m/z 1332, 53%) (Fig. S5 and S6†). The observed value (m/z 722) and isotropic pattern of this peak (Fig. S6†) is originated from the protonated form of complex 2 [Pt(ppy)(η1-S-SpyH)(PPh3)]+, which obviously establishes this complex in the gas phase. Moreover, the occurrence of the fragment ion at m/z 611 demonstrates that Spy ligand in complex 2 is labile and it easily undergoes Pt–S bond cleavage in the gas phase (refer to reactivity of complex 2 with HCl). This lability can be attributed to high trans influence of the σ Pt–C bond of ppy ligand which was observed recently in the cycloplatinated complexes having Pt(C^C*) moiety.28 The presence of a fragment peak at m/z 1332 suggests the uncoordinated nitrogen atom in thiolate ligand probably reacts with other reagents (see reactivity of complex 2 with HPF6). The low intensity peak (below 5%) at m/z 919 which is related to complex 1,2 may suggest that complex 2, through phosphine dissociation converts to complex 1 (see stability of complex 2 in solution). Other low intensity fragments at m/z 831, 1028, and 1070 could be assigned to [Pt(ppy)(η1-S-Spy)(PPh3)(pySH)]+, [{Pt(ppy)(μ2-Spy)}2(pySH)]+ and [{Pt(ppy)}2(PPh3) (μ2-Spy)]+, respectively (Fig. S5†).
The NMR spectroscopic data (typically in CD2Cl2) of complex 2 endorsed the formation of an isomer with the phosphine ligand and the nitrogen of ppy group in trans position. This feature is consistent with the transphobia effect (T),28–31 an expression that was previously agreed by several scientists to elucidate the stable geometry of square-planar d8 transition metal complexes. On the basis of T effect, the softer ancillary ligand, i.e. PPh3, must be located in cis position of Pt–C bond of cyclometalating ligand as the phosphorus atom has greater trans influence than the sulfur atom in thiolate group. The 31P {1H} NMR spectrum of complex 2 (Fig. S7†) includes a singlet resonance with platinum satellites (1JPtP = 4335 Hz). This high coupling constant value confirmed the trans disposition of the triphenylphosphine ligand with the Nppy atom.42,44 It also supports the higher trans influence of cyclometalated carbon respect to nitrogen. It is worthwhile mentioning that the proton NMR signals corresponding to both thiolate and ppy ligands in complex 2 are distinctly modified compared to complex 1 due to dissociation of Pt–Nspy bonds with the phosphine ligand. Three characteristic resonances (H2, H9 and H6′, the assignments were made on the basis of 2D experiment and schematic labeling is given in Fig. 2) are so sensitive to the coordination of PPh3 ligand. The H2 resonance in complex 2 appeared more deshielded42 (∼2.3 ppm) than complex 1, which is in agreement with the change of the ligand (from Spy to PPh3) in trans position of the nitrogen atom of ppy group in both complexes (Fig. 2). In complex 2, the H6′ peak did not display any coupling with the platinum center and it also indicated upfield shift (∼0.7 ppm). These results confirmed (in the substitution reaction) that the coordinated nitrogen atom of thiolate ligand is separated from metal center and this atom is not involved in the coordination sphere.45 The H9 signal in complex 2 in relation to complex 1 is shielded (∼0.6 ppm). This upfield shift, perhaps, can be attributed to an anisotropic shielding effect.28,46,47 It might have occurred by spatial proximity of the aromatic ring current of a phenyl group in the phosphine ligand with respect to the H9. This is consistent with C–H9⋯π interaction according to the cis embellishment of the Pt–P and Pt–Cppy bonds in complex 2. Moreover, H2 and H9 in complex 2 are coupled with phosphorous ligand and appeared as multiplet or doublet of doublets, respectively, as compared with complex 1.
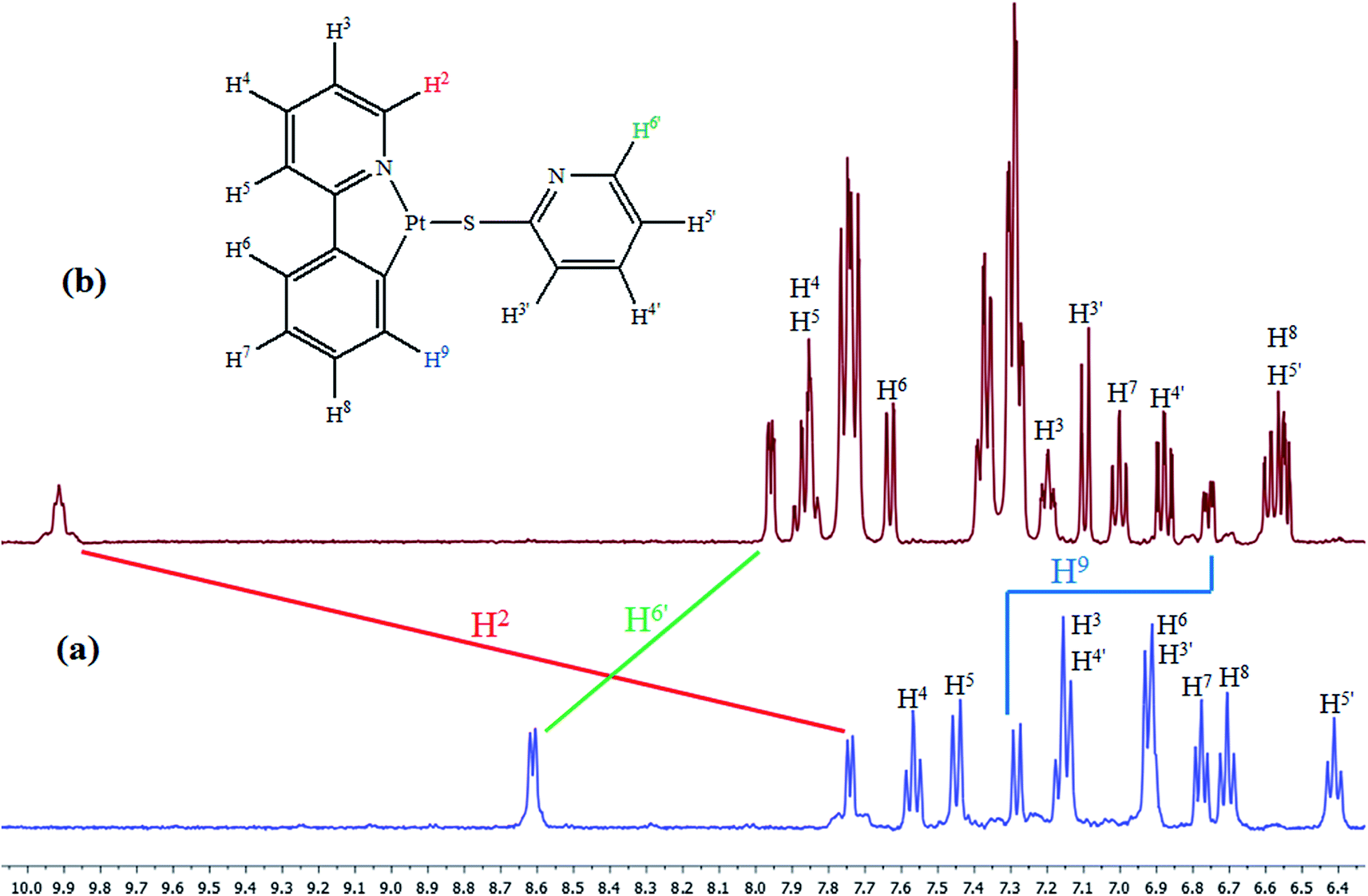 | ||
| Fig. 2 1H NMR spectra of complexes (a) 1 and (b) 2 in CD2Cl2 at room temperature. The signal assignments are depicted. | ||
Interestingly, complex 2 in dmso-d6 has experienced an equilibrium transformation over time (Fig. S8, S9† and Scheme 3 (step a)) and it experienced sluggish phosphine dissociation (detected by 31P {1H} NMR, Fig. S8b†). This observation suggests that perhaps an unstable intermediate [Pt(ppy)(η2-N,S-Spy)], I,33–35 (Scheme 3 (step a)) was formed during bond breaking. This intermediate probably has a weak intramolecular Pt–NSpy contact.20 This interaction is often labile in solution20 and easily broken by phosphine ligand and is converted back to complex 2. Furthermore, this suggests that the free nitrogen donor atom was necessary to cause this equilibrium because complex [Pt(ppy)(Sph)(PPh3)], 4, did not exhibit similar equilibrium.33 That is why complex 4 does not have pendant nitrogen in molecular structure (see structural determination). While this fluxional behavior occurred in hydrated (hydrous and aerated) solvent, these conditions caused the free phosphine easily oxidize to corresponding phosphine oxide (Fig. S8c–f† and Scheme 3).34 The side oxidation reaction stopped the equilibrium and it gradually transformed complex 2 to complex 1, perhaps by dimerization of two unstable intermediate I (Fig. S9, S10† and Scheme 3 (dimerization step)). This may support the idea that step (b) in Scheme 2 can be reversed under these circumstances. It is also worth mentioning that addition of excessive phosphorous ligand to the solution of complex 2 suppressed all above processes, which indicates that the weak Pt–NSpy contact in intermediate I is readily dissociated by PPh3.
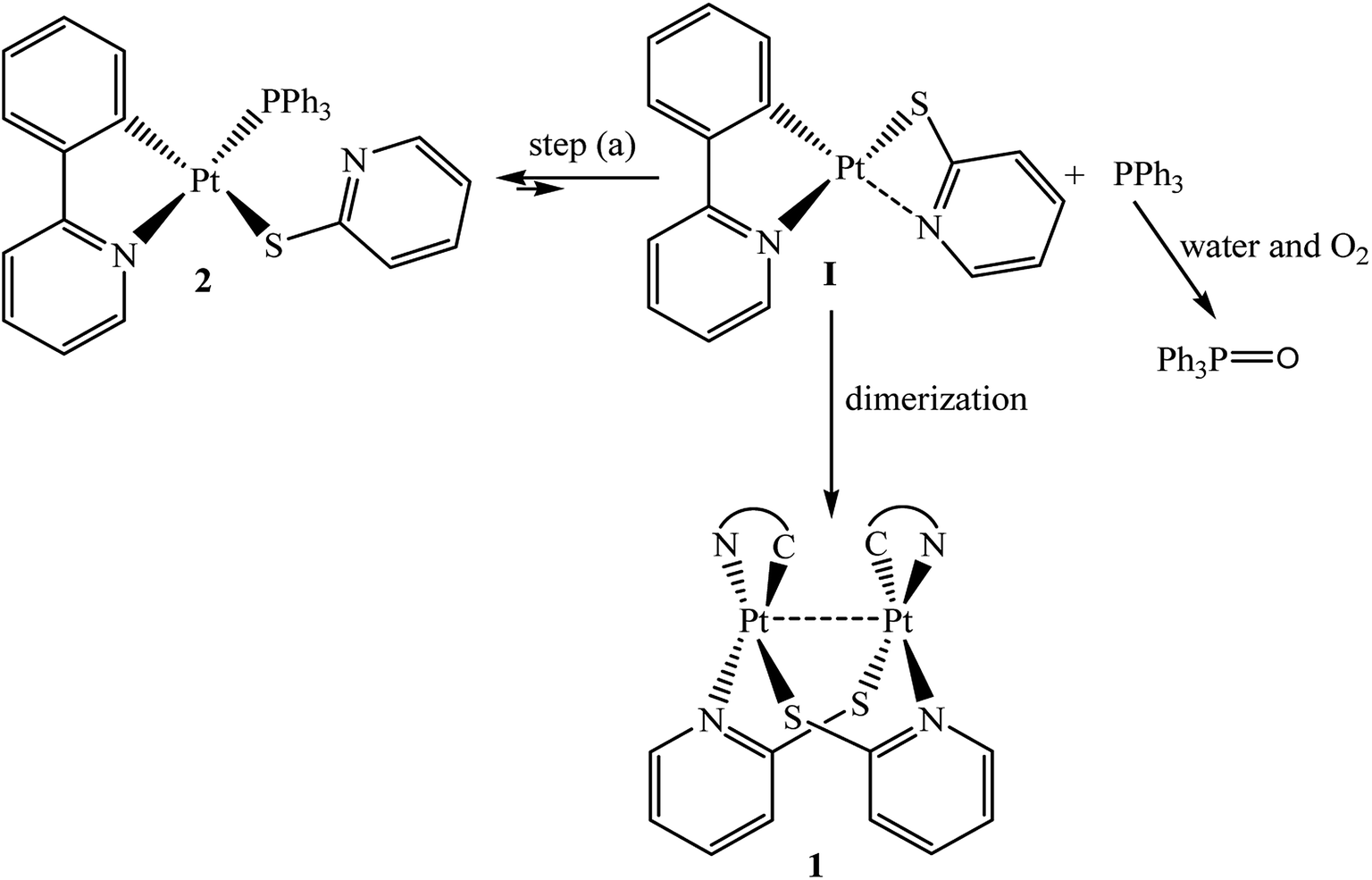 | ||
| Scheme 3 Equilibrium transformation and possible oxidation reaction for complex 2 in solution. | ||
Reaction of complex 2 with HX (X = Cl and PF6)
Complex 2 reacted very rapidly with HCl in stoichiometric ratio at room temperature. Remarkably, Pt–S bond was cleaved through the reaction (see Mass assignment of complex 2) and corresponding chloride complex 3 was formed with the loss of pySH ligand (Scheme 4).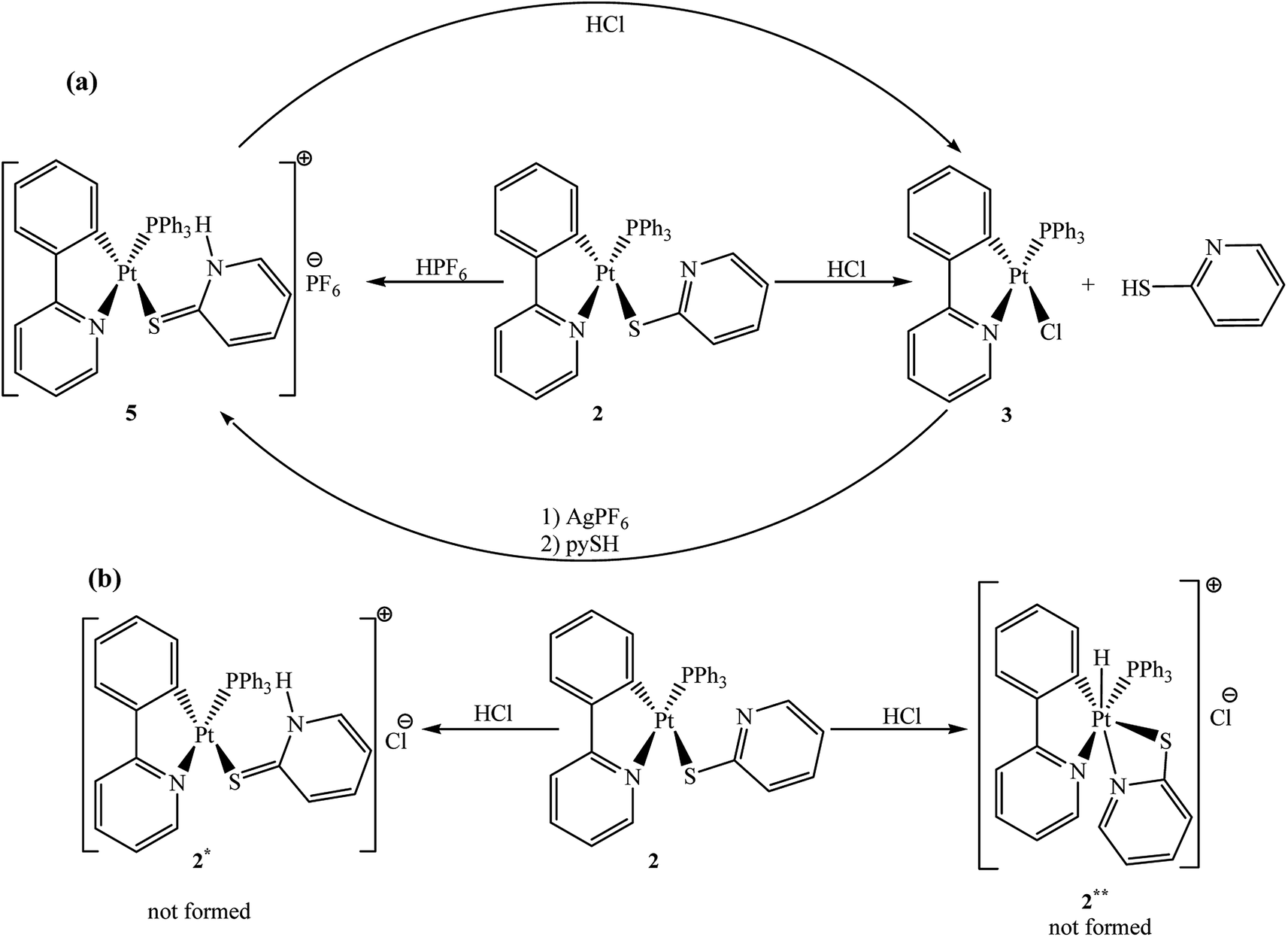 | ||
| Scheme 4 (a) Reaction of complex 2 with HCl and HPF6 at room temperature; (b) other potential products for reaction of complex 2 with HCl. | ||
The formation of these products in this condition suggested that step (b') in Scheme 2 can be inverted. Comparable product observed with complex 3, has already been published by Zucca and co-workers48,49 in the reaction of rollover complex [Pt(bipy-H)Me(DMSO)] with HCl, which gave the analogous chloride complex [Pt(bipy-H)Cl(DMSO)] and free methane.48 Under this condition, the protonation of the free nitrogen atom was not detected. Also, in the absence of nitrogen atom in complex 4, it reacted smoothly with HCl and rendered complex 3 and free thiophenol (PhSH).50 This might have occurred with the protonation process via an SE(ox) or an SE2 mechanism51–54 and probably uncoordinated nitrogen atom did not play an important role in the reaction.48,49 It should be noted that these two mechanisms are not relevant on present discussion.
Other possible routes for reaction of complex 2 with HCl are represented in Scheme 4. These routes are the protonation of the uncoordinated nitrogen atom of Spy ligand for preparation of the cationic adduct complex [Pt(ppy)(η1-S-Spy*)(PPh3)][Cl], 2*, or oxidation of platinum metal center in order to form Pt(IV) complex [Pt(ppy)(η2-N,S-Spy)(PPh3) (H)][Cl], 2**, but these proposed products were not formed (see below).
In the reaction of rollover complex [Pt(bipy-H)Me(DMSO)]48,49 with acidic species when coordinating anion (Cl−) was substituted with non-coordinating anion (BF4−), selective protonation occurred on the free nitrogen atom and platinum center remained intact.48,49,55 Thus, in the above protonation reaction (Scheme 4), reaction with HPF6 containing uncoordinating PF6− anion, caused the protonation of pendant nitrogen atom. When complex 2 treated with HPF6, complex [Pt(ppy)(η1-S-Spy*)(PPh3)][PF6], 5, was yielded in which Spy* ligand can be envisaged as a tautomeric form of the neutral thiol ligand. Therefore, it demonstrates that the formation of complex 2* (Scheme 4) requires a non-coordinating anion.48
Complex 5 was characterized by multinuclear (1H and 31P {1H}) NMR spectroscopy in solution. The 31P {1H} NMR spectrum of complex 5 (Fig. S11†) indicated two different phosphorus resonances. The first signal appeared at δ = 22.3 ppm as a singlet resonance flanked by platinum satellites (1JPtP = 4156 Hz) which was assigned to PPh3 ligand. It is well known that coupling constant is a good indicator of the kind of atom in trans position of phosphorus atom. Therefore, this large coupling constant supports a P–Pt–Nppy trans arrangement in complex 5, indicating a good agreement with coupling constant that was found for complex 2, only less by ca. 190 Hz. This reduction can be attributed to the cis influence of the N-protonated 2-thiolate fragment which is considerably increased compared to that of the 2-thiolate ligand.56–61 Similar behavior was previously reported for complex trans-[PtBr(C5H5N-C2)(PPh3)2] in reaction with HClO4.58 The second signal at δ = −144.3 was assigned to PF6 counter-anion which is coupled to fluorine nucleus to give a septet and was separated by 708 Hz. In the 1H NMR spectrum, a broad signal was detected at down field (δ = 13.11 ppm) which is related to N–H proton (Fig. S12†),48,57,58 and it disappeared immediately upon treatment with D2O (Fig. S13†).57 The protonation of the nitrogen atom led all protons of Spy ligand shift to down field of about 0.2–0.9 ppm, as anticipated from the existence of a positive charge.49,57 With the reduction of trans influence of protonated thiolate ligand, coupling constant value of H9 was increased by ca. 8 Hz.
Templeton, et al. reported an interesting protonation reaction for the preparation of platinum–hydride complexes.62 In this reaction, κ2 complexes [(κ2–TtR)PtMe2], which include a free nitrogen, were treated with a strong acid (HBF4) and yielded κ3 complexes [(κ3–TtR)PtMe2(H)]BF4. In these Pt–H complexes all three nitrogens were coordinated to the platinum center and produced two stable chelates. In the case of reaction of complex 2 with HCl, if complex 2** was formed as a presumptive product, the thiolate ligand in complex 2** acts as a four membered chelate (η2-N,S-Spy). The chelates formed by 2-N,S-thiolate ligands are intrinsically strained20 and perhaps this structural strain prevented formation of complex 2** (Scheme 4).
Complex 5 can be obtained in another synthetic route involving treatment of complex 3 with AgPF6 (in acetonitrile solvent) to abstract the chloride ligand from the platinum center as AgCl precipitate which is separable from solution by filtration. Subsequently, pyridine-2-thiol was added to the resulting solution to form complex 5. Moreover, addition of equimolecular amount of HCl to a solution of complex 5 rendered complex 3 (Scheme 4).
Structural determination
Single-crystal X-ray diffraction investigation was carried out on complex 4 to approve its molecular structure. Crystallographic data is collected in Table S3,† and the perspective drawing of this complex is illustrated in Fig. 3. | ||
| Fig. 3 Representations of the X-ray crystal of complex [Pt(ppy)(Sph)(PPh3)], 4, showing all non-hydrogen atoms as 40% thermal ellipsoids. Pt1–C1 2.065(12), Pt1–N1 2.073(9), Pt1–P1 2.244(3), Pt1–S1 2.361(3), C1–Pt1–N1 81.9(4), C1–Pt1–P1 97.2(3), C1–Pt1–S1 169.5(3), N1–Pt1–P1 178.2(3), N1–Pt1–S1 90.2(3), P1–Pt1–S1 90.95(11). | ||
Complex 4 reveals a distorted square planar coordination environment around Pt(II) center. This distortion is due to small bite angle [81.9(4)°] of the ppy cyclometalated ligand. This narrow bite angle with bond distances for cyclometalated chelate (i.e. Pt–Cppy and Pt–Nppy) are comparable to those observed in other five membered rings of 2-phenylpyridinate platinum(II) complexes.23,43,63,64 In this structure, the metalated carbon atom of the C^N ligand accommodates the coordinated phosphine ligand in cis arrangement of Pt–Cppy bond, as a result of high trans influence of carbon atom.44 Therefore, PPh3 ligand is located trans to nitrogen atom of ppy ligand and bond distance of the Pt–P is similar to those observed in complex [Pt(ppy)(PTA)Cl]42 and notably shorter than platinum–phosphorous bond lengths, positioned trans to carbon atom of cyclometalated ligand.63 Also, sulfur ligand completes the coordination sphere of platinum atom. The Pt–SPh distance is normal and fall in the typical range for cycloplatinated(II) complexes containing ppy moieties with those positioned trans to the σ-bound carbon atoms. The Pt–C metalated bond lengths is noticeably changed according to the trans influence of the SPh ligand occupying its trans site.28
The molecular structure of complex 4 reveals that the thiolate ligand is bent toward phosphine ligand (Fig. 3). The similar orientation has already been observed in (arylthiolato) palladium complexes with thiolate ligands.65
Additional examination on the molecular packing of the crystal structure of complex 4 displayed the presence of intramolecular interactions (see below) such as C–H⋯π interaction, π⋯π stacking, without any notable Pt⋯Pt contact. This molecular structure reveals a moderate short C–H⋯π intramolecular interaction (Fig. S14†) between the hydrogen atom adjacent to coordinated carbon atom of cyclometalated ligand and the phenyl ring of the triphenylphosphine ligand (C–H⋯π; C2⋯Cph (PPh3) = 3.312 Å). This interaction has caused the coordinated hydrogen atom of ppy ligand to be more shielded in NMR spectroscopy (Fig. 2).28
In complex 4, the aromatic ring of the thiolate ligand exhibits weak intramolecular π⋯π interaction with one of the phenyl groups of the PPh3 ligand and the distance between two centroids is 3.837 Å (Fig. S15†), which are located nearly parallel to each other (15.6(7)° angle between two aromatic rings).
Conclusions
The half-lantern platinum(II) complex 1 is a useful precursor for substitution reaction with triphenylphosphine ligand. This process proceeded via bond dissociation between platinum and nitrogen atom of thiolate bridging ligand and yielded platinum(II) complex 2, which is comprised of the free pyridyl group. Also, the transphobia effect directs the entering phosphine donor ligand to cis position of carbon atom of cyclometalating fragment, as concluded from spectroscopic data.Complex 2 underwent protonation reaction and the type of anion played an important role in the outcome of the protonation reaction. Thus, its reaction with an acid involving a coordinating anion such as chloride (Cl−), proceeded through the cleavage of the Pt–Spy bond and formation of complex 3. In contrast, reaction of complex 2 with an acid containing an uncoordinating anion such as PF6−, proceeded by the protonation of the pendant nitrogen atom and formation of complex 5 as a cationic species.
Experimental section
General procedures and materials
All NMR spectra (1H and 31P {1H}) were recorded on a Brucker Avance DPX 400 MHz instrument and are referenced to the residual peak of the solvent, i.e. CD2Cl2, CDCl3, dmso-d6 and acetone-d6, the chemical shifts (δ) being reported as ppm and coupling constants (J) expressed in Hz. The microanalyses were performed using a vario EL CHNS elemental analyzer. Electrospray ion mass spectrum (ESI-MS) was recorded by a HP-5989B spectrometer using methanol–water as the mobile phase. All solvents were purified and dried according to standard procedures.66 2-Phenylpyridine (ppyH), hexafluorophosphoric acid solution (HPF6), silver hexafluorophosphate (AgPF6), pyridine-2-thiol (pySH) and thiophenol (PhSH) were purchased from Aldrich or Acros. The complexes [Pt(ppy)(DMSO)(Cl)], A,43 [{Pt(ppy)(μ2-Spy)}2], 1,3,32 [{Pt(ppy)(μ2-Spy)Cl}2], 1a,3 [Pt(ppy)(PPh3)Cl], 3,41,42 were prepared as reported in literature. The NMR labeling are shown in Fig. 2 for clarifying the chemical shift assignments. Additional data for complex 3: 1H NMR (400 MHz, dmso-d6, 20 °C, δ): 9.67 (m, 3JPtH = not resolved, 1H, H2), 8.20–8.13 (m, 2H, H4, H5), 7.75 (dd, 3JHH = 7.9, 4JHH = 0.9, 1H, H6), 7.71–7.66 (m, 6H, Ho of PPh3), 7.59–7.55 (m, 1H, H3), 7.52–7.43 (m, 9H, Hp and Hm of PPh3), 6.95 (td, 3JHH = 8.0, 4JHH = 1.2, 1H, H7), 6.59–6.46 (m, 2H, H8, H9). 31P {1H} NMR (162 MHz, dmso-d6, 20 °C, δ): 22.8 (s, 1JPtP = 4341, 1P). 1H NMR (400 MHz, CD2Cl2, 20 °C, δ): 9.82 (m, 3JPtH = not resolved, 1H, H2), 7.93 (td, 3JHH = 8.1, 4JHH = 1.6, 1H, H4) 7.85 (d, 3JHH = 8.1, 1H, H5), 7.82–7.76 (m, 6H, Ho of PPh3), 7.56 (dd, 3JHH = 7.8, 4JHH = 1.3, 1H, H6), 7.49–7.32 (m, 10H, H3, Hp and Hm of PPh3), 6.96 (td, 3JHH = 7.8, 4JHH = 1.1, 1H, H7), 6.66 (dd, 3JPtH = 53.6, 3JHH = 7.8, 4JPH = 2.2, 1H, H9), 6.51 (td, 3JHH = 7.8, 4JHH = 1.2, 1H, H8). 31P {1H} NMR (162 MHz, CD2Cl2, 20 °C, δ): 23.358 (s, 1JPtP = 4338, 1P).[Pt(ppy)(η1-S-Spy)(PPh3)], 2
[Pt(ppy)(SPh)(PPh3)], 4
Complex 3 (100 mg, 0.15 mmol) was added to an ethanolic solution of sodium phenylthiolate ligand (NaC6H5S) under inert atmospheric condition [NaC6H5S prepared by dissolving of sodium (4.6 mg, 0.20 mmol) in 10 mL of absolute ethanol and is treated with thiophenol (15.3 μL, 0.15 mmol)]. Bright orange solution was formed and after stirring for 5 hours at room temperature an orange solid precipitated which was separated, washed with ethanol (3 × 2 mL) and cold acetone (2 × 2 mL) and dried to obtain complex 4. Yield: 57 mg, 53%; mp 210 °C. Elem. anal. calcd for C35H28NPPtS (720.14): C, 58.32; H, 3.92; N, 1.94; found: C, 58.16; H, 3.87; N, 1.98. 1H NMR (400 MHz, dmso-d6, 20 °C, δ): 9.81 (m, 3JPtH = not resolved, 1H, H2), 8.14 (d, 3JHH = 8.2, 1H, H5), 8.05 (td, 3JHH = 8.2, 4JHH = 1.4, 1H, H4), 7.80 (d, 3JHH = 7.8, 1H, H6), 7.67–7.62 (m, 6H, Ho of PPh3), 7.46–7.34 (m, 10H, H3, Hp and Hm of PPh3), 7.15 (dd, 3JHH = 8.3, 4JHH = 1.2, 2H, H2′), 6.98 (td, 3JHH = 7.8, 4JHH = 1.1, 1H, H7), 6.75 (t, 3JHH = 7.9, 2H, H3′), 6.68–6.65 (m, 2H, H9, H4′), 6.57 (td, 3JHH = 7.8, 4JHH = 1.1, 1H, H8). 31P {1H} NMR (162 MHz, dmso-d6, 20 °C, δ): 23.5 (s, 1JPtP = 4295, 1P). 1H NMR (400 MHz, acetone-d6, 20 °C, δ): 10.05 (dddd, 3JPtH = 31.2, 3JHH = 5.7, 4JHH = 1.6, 5JHH = 0.7, 4JPH = 3.9, 1H, H2), 8.11–8.03 (m, 2H, H4, H5), 7.81–7.73 (m, 7H, H6, Ho of PPh3), 7.44 (tdd, 3JHH = 7.4, 4JHH = 1.4, 5JPH = 2.0, 3H, Hp of PPh3), 7.37–7.31 (m, 7H, H3, Hm of PPh3), 7.23 (ddd, 3JHH = 8.3, 4JHH = 1.1, 5JPH = 3.4, 2H, H2′), 6.99 (td, 3JHH = 7.8, 4JHH = 1.2, 1H, H7), 6.81 (ddd, 3JPtH = 42.7,3JHH = 7.9, 4JHH = 0.9, 4JPH = 3.2, 1H, H9), 6.72 (t, 3JHH = 7.8, 2H, H3′), 6.66 (td, 3JHH = 7.7, 4JHH = 1.2, 1H, H4′), 6.57 (td, 3JHH = 7.7, 4JHH = 1.2, 1H, H8). 31P {1H} NMR (162 MHz, acetone-d6, 20 °C, δ): 23.9 (s, 1JPtP = 4301, 1P). 1H NMR (400 MHz, CD2Cl2, 20 °C, δ): 9.96 (m, 3JPtH = 31.1, 1H, H2), 7.89–7.81 (m, 2H, H4, H5), 7.75–7.71 (m, 6H, Ho of PPh3), 7.63 (dd, 3JHH = 7.8, 4JHH = 1.1, 1H, H6), 7.42–7.38 (m, 3H, Hp of PPh3), 7.34–7.25 (m, 8H, H2′ and Hm of PPh3), 7.16–7.13 (m, 1H, H4′), 7.01(td, 3JHH = 7.4, 4JHH = 1.1, 1H, H3), 6.83–6.69 (m, 4H, H7, H9, H3′), 6.60 (td, 3JHH = 7.7, 4JHH = 1.4, 1H, H8). 31P {1H} NMR (162 MHz, CD2Cl2, 20 °C, δ): 23.4 (s, 1JPtP = 4304, 1P).[Pt(ppy)(η1-S-Spy*)(PPh3)][PF6], 5
Reaction of complexes 2 and 5 with HCl
To a solution of complexes 2 or 5 (0.14 mmol) in acetone (20 mL) with vigorous stirring was added HCl (0.14 mmol, 1 mL of aqueous 0.05 M) and after 3 h the solution was concentrated to a small volume (5 mL). The yellow solution was extracted with CH2Cl2 (10 mL) and dried with MgSO4, and concentrated to a small volume (1 mL). Then, n-hexane (10 mL) was added to precipitate complex 3 (confirmed by NMR spectroscopy) as a yellow solid and dried under vacuum.Acknowledgements
This work was supported by the Institute for Advanced Studies in Basic Sciences (IASBS) Research Council and the Iran National Science Foundation (Grant no. 93026027). Technical support of the Chemistry Computational Center at Shahid Beheshti University is gratefully acknowledged. Thanks are also due to Dr A. Neshat, IASBS, for valuable suggestions, Mr A. Biglari, the operator of Bruker NMR instrument at IASBS, for recording the NMR spectra and Prof. El5ena Lalinde, Universidad de La Rioja, for ESI-MS measurements. This paper is dedicated to Professor S. Masoud Nabavizadeh on the occasion of his 45th birthday.References
- V. Sicilia, P. Borja and A. Martín, Inorganics, 2014, 2, 508 CrossRef CAS.
- R. Aoki, A. Kobayashi, H.-C. Chang and M. Kato, Bull. Chem. Soc. Jpn., 2011, 84, 218–225 CrossRef CAS.
- T. Koshiyama, A. Omura and M. Kato, Chem. Lett., 2004, 33, 1386–1387 CrossRef CAS.
- K. Saito, Y. Nakao, K. Umakoshi and S. Sakaki, Inorg. Chem., 2010, 49, 8977–8985 CrossRef CAS PubMed.
- E. A. Katlenok, A. A. Zolotarev, A. Y. Ivanov, S. N. Smirnov, R. I. Baichurin and K. P. Balashev, Russ. J. Coord. Chem., 2015, 41, 387–394 CrossRef CAS.
- B. Ma, P. I. Djurovich, S. Garon, B. Alleyne and M. E. Thompson, Adv. Funct. Mater., 2006, 16, 2438–2446 CrossRef CAS.
- V. Sicilia, P. Borja, J. M. Casas, S. Fuertes and A. Martín, J. Organomet. Chem., 2013, 731, 10–17 CrossRef CAS.
- V. Sicilia, J. Forniés, J. M. Casas, A. Martín, J. A. López, C. Larraz, P. Borja, C. Ovejero, D. Tordera and H. Bolink, Inorg. Chem., 2012, 51, 3427–3435 CrossRef CAS PubMed.
- X. Qi, P. I. Djurovich, N. C. Giebink, M. E. Thompson and S. R. Forrest, Chem. Phys. Lett., 2008, 458, 323–328 CrossRef CAS.
- V. Sicilia, P. Borja, M. Baya and J. M. Casas, Dalton Trans., 2015, 44, 6936–6943 RSC.
- V. Sicilia, M. Baya, P. Borja and A. Martín, Inorg. Chem., 2015, 54, 7316–7324 CrossRef CAS PubMed.
- T. Koshiyama and M. Kato, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, m173–m176 Search PubMed.
- Z. Wang, L. Jiang, Z.-P. Liu, C. R. R. Gan, Z. Liu, X.-H. Zhang, J. Zhao and T. S. A. Hor, Dalton Trans., 2012, 41, 12568–12576 RSC.
- A. Rodríguez, M. J. Romero, A. Fernández, M. López-Torres, D. Vázquez-García, L. Naya, J. M. Vila and J. J. Fernández, Polyhedron, 2014, 67, 160–170 CrossRef.
- M. J. Romero, A. Rodríguez, A. Fernández, M. López-Torres, D. Vázquez-García, J. M. Vila and J. J. Fernández, Polyhedron, 2011, 30, 2444–2450 CrossRef CAS.
- M. A. Bennett, S. K. Bhargava, E. C.-C. Cheng, W. H. Lam, T. K.-M. Lee, S. H. Privér, J. Wagler, A. C. Willis and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 7094–7103 CrossRef CAS PubMed.
- S. J. Hoseini, S. M. Nabavizadeh, S. Jamali and M. Rashidi, J. Organomet. Chem., 2007, 692, 1990–1996 CrossRef.
- S. J. Hoseini, H. Nasrabadi, S. M. Nabavizadeh and M. Rashidi, J. Organomet. Chem., 2013, 725, 22–27 CrossRef CAS.
- E. S. Raper, Coord. Chem. Rev., 1997, 165, 475–567 CrossRef CAS.
- E. S. Raper, Coord. Chem. Rev., 1996, 153, 199–255 CrossRef CAS.
- E. S. Raper, Coord. Chem. Rev., 1985, 61, 115–184 CrossRef CAS.
- J. R. Berenguer, E. Lalinde, A. Martin, M. Teresa Moreno, S. Ruiz, S. Sanchez and H. R. Shahsavari, Chem. Commun., 2013, 49, 5067–5069 RSC.
- J. R. Berenguer, E. Lalinde, A. Martín, M. T. Moreno, S. Ruiz, S. Sánchez and H. R. Shahsavari, Inorg. Chem., 2014, 53, 8770–8785 CrossRef CAS PubMed.
- C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC.
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed.
- S. Miranda, E. Vergara, F. Mohr, D. de Vos, E. Cerrada, A. Mendía and M. Laguna, Inorg. Chem., 2008, 47, 5641–5648 CrossRef CAS PubMed.
- E. Guerrero, S. Miranda, S. Lüttenberg, N. Fröhlich, J.-M. Koenen, F. Mohr, E. Cerrada, M. Laguna and A. Mendía, Inorg. Chem., 2013, 52, 6635–6647 CrossRef CAS PubMed.
- S. Fuertes, A. J. Chueca and V. Sicilia, Inorg. Chem., 2015, 54, 9885–9895 CrossRef CAS PubMed.
- M.-J. Oliva-Madrid, J.-A. García-López, I. Saura-Llamas, D. Bautista and J. Vicente, Organometallics, 2014, 33, 33–39 CrossRef CAS.
- J. Vicente, J.-A. Abad, E. Martínez-Viviente and P. G. Jones, Organometallics, 2002, 21, 4454–4467 CrossRef CAS.
- M. Crespo, J. Granell, X. Solans and M. Font-Bardia, J. Organomet. Chem., 2003, 681, 143–149 CrossRef CAS.
- M. Dadkhah Aseman, S. M. Nabavizadeh, H. R. Shahsavari and M. Rashidi, RSC Adv., 2015, 5, 22692–22702 RSC.
- Y. Nakatsu, Y. Nakamura, K. Matsumoto and S. i. Ooi, Inorg. Chim. Acta, 1992, 196, 81–88 CrossRef CAS.
- T. S. Lobana, R. Verma and A. Castineiras, Polyhedron, 1998, 17, 3753–3758 CrossRef CAS.
- S. Narayan and V. Jain, Transition Met. Chem., 1999, 24, 409–413 CrossRef CAS.
- C. R. Hilliard, N. Bhuvanesh, J. A. Gladysz and J. Blumel, Dalton Trans., 2012, 41, 1742–1754 RSC.
- T. E. Barder and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 5096–5101 CrossRef CAS PubMed.
- S. A. Buckler, J. Am. Chem. Soc., 1962, 84, 3093–3097 CrossRef CAS.
- S. M. Nabavizadeh, H. R. Shahsavari, M. Namdar and M. Rashidi, J. Organomet. Chem., 2011, 696, 3564–3571 CrossRef CAS.
- H. Honda, T. Matsumoto, M. Sakamoto, A. Kobayashi, H.-C. Chang and M. Kato, Inorg. Chem., 2013, 52, 4324–4334 CrossRef CAS PubMed.
- G. L. Edwards, D. S. C. Black, G. B. Deacon and L. P. G. Wakelin, Can. J. Chem., 2005, 83, 980–989 CrossRef CAS.
- H. Samouei, M. Rashidi and F. W. Heinemann, J. Iran. Chem. Soc., 2014, 11, 1207–1216 CrossRef CAS.
- A. Esmaeilbeig, H. Samouei, S. Abedanzadeh and Z. Amirghofran, J. Organomet. Chem., 2011, 696, 3135–3142 CrossRef CAS.
- A. Díez, J. Forniés, A. García, E. Lalinde and M. T. Moreno, Inorg. Chem., 2005, 44, 2443–2453 CrossRef PubMed.
- G. Cervantes, S. Marchal, M. J. Prieto, J. M. Pérez, V. M. González, C. Alonso and V. Moreno, J. Inorg. Biochem., 1999, 77, 197–203 CrossRef CAS PubMed.
- J. Forniés, V. Sicilia, C. Larraz, J. A. Camerano, A. Martín, J. M. Casas and A. C. Tsipis, Organometallics, 2010, 29, 1396–1405 CrossRef.
- V. Sicilia, S. Fuertes, A. Martín and A. Palacios, Organometallics, 2013, 32, 4092–4102 CrossRef CAS.
- A. Zucca, G. L. Petretto, S. Stoccoro, M. A. Cinellu, M. Manassero, C. Manassero and G. Minghetti, Organometallics, 2009, 28, 2150–2159 CrossRef CAS.
- L. Maidich, G. Zuri, S. Stoccoro, M. A. Cinellu, M. Masia and A. Zucca, Organometallics, 2013, 32, 438–448 CrossRef CAS.
- R. Zanella, R. Ros and M. Grazian, Inorg. Chem., 1973, 12, 2736–2738 CrossRef CAS.
- E. W. Kalberer, J. F. Houlis and D. M. Roddick, Organometallics, 2004, 23, 4112–4115 CrossRef CAS.
- M. Golbon Haghighi, S. M. Nabavizadeh, M. Rashidi and M. Kubicki, Dalton Trans., 2013, 42, 13369–13380 RSC.
- G. Mazzone, N. Russo and E. Sicilia, Inorg. Chem., 2011, 50, 10091–10101 CrossRef CAS PubMed.
- H. R. Shahsavari, M. Rashidi, S. M. Nabavizadeh, S. Habibzadeh and F. W. Heinemann, Eur. J. Inorg. Chem., 2009, 3814–3820 CrossRef CAS.
- L. Maidich, G. Zuri, S. Stoccoro, M. A. Cinellu and A. Zucca, Dalton Trans., 2014, 43, 14806–14815 RSC.
- L. Rigamonti, C. Manassero, M. Rusconi, M. Manassero and A. Pasini, Dalton Trans., 2009, 1206–1213 RSC.
- B. Crociani, F. di Bianca, A. Giovenco and A. Scrivanti, J. Organomet. Chem., 1983, 251, 393–411 CrossRef CAS.
- B. Crociani, F. Di Blanca, A. Giovenco, A. Berton and R. Bertani, J. Organomet. Chem., 1989, 361, 255–267 CrossRef CAS.
- P. G. Waddell, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2010, 39, 8620–8625 CAS.
- L. Rigamonti, M. Rusconi, C. Manassero, M. Manassero and A. Pasini, Inorg. Chim. Acta, 2010, 363, 3498–3505 CrossRef CAS.
- L. Rigamonti, M. Rusconi, A. Forni and A. Pasini, Dalton Trans., 2011, 40, 10162–10173 RSC.
- B. E. Frauhiger, P. S. White and J. L. Templeton, Organometallics, 2012, 31, 225–237 CrossRef CAS.
- P. Hamidizadeh, M. Rashidi, S. M. Nabavizadeh, M. Samaniyan, M. Dadkhah Aseman, A. M. Owczarzak and M. Kubicki, J. Organomet. Chem., 2015, 791, 258–265 CrossRef CAS.
- Á. Díez, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2011, 255, 2426–2447 CrossRef.
- L. Munjanja, W. W. Brennessel and W. D. Jones, Organometallics, 2015, 34, 4574–4580 CrossRef CAS.
- B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, Longman Scientific & Technical, 5th edn, 1989 Search PubMed.
- Stoe & Cie, X–AREA: Program for the Acquisition and Analysis of Data, Version 1.30, Stoe & Cie GmbH: Darmatadt, Germany, 2005 Search PubMed.
- Stoe & Cie, X–RED: Program for Data Reduction and Absorption Correction, Version 1.28b, Stoe & Cie GmbH: Darmatadt, Germany, 2005 Search PubMed.
- Stoe & Cie, X–SHAPE: Program for Crystal Optimization for Numerical Absorption Correction, Version 2.05, Stoe & Cie GmbH: Darmatadt, Germany, 2004 Search PubMed.
- G. M. Sheldrick, SHELX97. Program for Crystal Structure Solution, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELX97. Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- E. Prince, International Tables for X-ray Crystallography, Vol C, Kluwer Academic Publisher, Doordrecht, The Netherlands, 1995 Search PubMed.
- F. H. Allen, O. Johnson, G. P. Shields, B. R. Smith and M. Towler, J. Appl. Crystallogr., 2004, 37, 335–338 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van der Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- M. N. Burnett and C. K. Johnson, ORTEP-III Report ORNL-6895. Oak Ridge National Laboratory, Tennessee, USA, 1996 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. Coppens, L. Leiserowitz and D. Rabinovich, Acta Crystallogr., 1965, 18, 1035–1038 CrossRef CAS.
- Stoe & Cie, X-STEP32: Crystallographic Package, Version 1.07b, Stoe & Cie GmbH, Darmstadt, Germany, 2000 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, 2010 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full NMR spectra, crystallographic and computational details. CCDC 1457811. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra15604e |
| This journal is © The Royal Society of Chemistry 2016 |