
Reactor production of no-carrier-added 199Au for biomedical applications
K. V. Vimalnath,
Sudipta Chakraborty and
Ashutosh Dash*
Isotope Production and Applications Division, Bhabha Atomic Research Centre, Mumbai, India. E-mail: adash@barc.gov.in; Fax: +91-22-25505151; Tel: +91-22-25595372
First published on 25th August 2016
Abstract
This investigation described development of a technology for the reactor production of no-carrier-added (NCA) 199Au through neutron activation of natural Pt in a research reactor followed by chemical separation of 199Au employing liquid–liquid extraction (LLX) technique using ethyl acetate. A thorough optimization of experimental parameters was carried out primarily to arrive at the separation conditions resulting in quantitative extraction of 199Au into the organic phase. The reported procedure has been found to be prolific in providing 199Au with >95% yields with acceptable radionuclidic and radiochemical purity. An optimized radiochemical processing procedure for obtaining NCA 199Au of requisite purity in high yields suitable for clinical use has been the positive outcome.
Introduction
Gold-199 [T1/2 = 3.14 d, Eβmax = 244 keV (21.5%), 294 keV (72%) and 452 keV (6.5%), Eγ = 158.4 keV (36.9%), 208.2 keV (8.4%)] is a promising therapeutic radionuclide that has attracted considerable attention by virtue of its favorable chemical and nuclear decay characteristics.1–3 The 294 keV β− particle emitted by 199Au has a maximum tissue penetration range of 0.8 mm and its average β− particle energy, 84 keV, has a mean free path in tissue of 100 μm, making this radionuclide ideal for delivering energy to small volumes, including micro-metastatic disease, and tumor cells near the surface of cavities and is therefore well suited for radioimmunotherapy.4 The emission of accompanying gamma photons of 158.4 keV of 36.9% yield offers the scope for imaging the biodistribution and excretion kinetics with the same radiolabeled preparation used for therapy, and allows dosimetry to be performed before and during treatment as well. The 3.14 day half-life of 199Au offers extended time periods to radiolabel and purify 199Au labeled radiopharmaceuticals, provides logistical advantage for shipment to sites distant from the production facility in a clinically relevant time frame, and is compatible with the temporal accumulation of antibodies in tumors.4,5 Interest in the use of 199Au was spun primarily due to its ability to form clusters of 11 gold atoms, which could then attach to monoclonal antibodies that selectively recognizes and binds on target cells.5–9 Gold nanoparticles labeled with 199Au offers the scope of using as a probe for single photon emission computed tomography (SPECT) imaging to study the biodistribution and clearance of agents as well as to assess the dosimetry and maximum tolerated dose (MTD) of therapeutic gold agents.10,11While the radionuclide 199Au harbor significant therapeutic potential4–19 and is poised to take radionuclide therapy to a leap forward, cost effective availability of desired quantities and qualities represents the key determinant that underpin its survival, strength and success. In view of this, acquiring local production capability of 199Au of desired quantities and qualities seemed not only an attractive prospect to meet the local demands but also to empower future development and thus pursued. In this premise, it is imperative to consider all possible 199Au production options conscientiously. This radionuclide can be produced either using the reactor20–27,38,39 or accelerator path.28–33 In the quest for a viable 199Au production route, our attention was turned towards reactor path. Both the “direct” and “indirect” reactor production routes can be followed to obtain 199Au. While reactor production following double neutron capture on 197Au [197Au(n,γ)198Au(n,γ)199Au] is the least intricate route and generate negligible amount of radioactive waste, the resulting product is a mixture of 198Au and 199Au with low specific activity. In light of the perceived need to use 199Au for radioimmunotherapy (RIT), use of NCA 199Au constitutes a necessity owing to the limited concentrations of antigen expressed on the tumor cell surface. In this premise, development of an indigenous indirect reactor production route consisting of  is not only an interesting prospect owing to its ability to provide NCA 199Au, but is viewed as a necessity.
is not only an interesting prospect owing to its ability to provide NCA 199Au, but is viewed as a necessity.
With a view to separate 199Au from the neutron irradiated 198Pt target, two chemical separation strategies including liquid–liquid extraction (LLX) and ion-exchange separation method are widely followed.5,27,34–41 In the quest for a viable method to undertake separation of 199Au from bulk of Pt, our attention was turned towards the use of LLX method owing to operational simplicity, versatility, ready adaptability for remote operation and flexibility to scaling up or down to its level of operation in response to requirements. While the LLX method obviously hold promise as an attractive approach, selection of an extractant to isolate 199Au of requisite purity represents a key determinant in ensuring successful implementation of this strategy.
A general survey of the literature reveals that the extractants 1-phenyl-3-methyl-4-trifluoroacetyl-pyrazolone-5,35 di-(2-ethylhexyl)phosphoric acid (HDEHP)38 and trioctylamine (TOA)39 have been exploited for the isolation of 199Au from bulk of Pt. We have chosen ethyl acetate as the extractant owing to its ready availability of requisite purity at low cost, relatively non-toxic, non-hygroscopic and volatile. The prospect of using ethyl acetate seemed appealing as it offers the scope of recovering 199Au by subsequent removal of the extractant by distillation. In order to tap the potential of ethyl acetate in the isolation of 199Au from bulk of Pt, a careful scrutiny of the distribution ratios of 199Au and Pt was hence considered worthwhile investigating to arrive at the optimum separation conditions and thus pursued diligently.
Herein, we report a systematic approach consisting of a number of sequential steps that contributed to the development of an indigenous processing method for the production of NCA 199Au of requisite purity and yield for clinical applications. Since the inherent success of a 199Au production scheme requires a thorough knowledge of all the pertinent key factors, the issues underlying neutron irradiation parameters, process equipment design and radiochemical separation procedure optimization to isolate NCA 199Au were adequately addressed. We described the overall process, an overview of our experience and quality evaluation of 199Au.
Experimental
Materials
Platinum sponge targets (natural isotopic composition) spec pure grade procured from by M/s. Roviur International, Singapore was used as target for neutron irradiation in the reactor. Ethyl acetate was procured from Sigma-Aldrich India, Mumbai. All chemicals and reagents used in this work were of analytical grade and were procured from Merck India Ltd, Mumbai, India or BDH Industries Limited, Mumbai, India. Paper chromatography (PC) strips were purchased from Whatman, UK. The glass equipments required for the radiochemical separations were designed and fabricated in-house.Equipments
A high resolution γ-ray spectrometer consisting of HPGe detector coupled to a PC-based 4 K channel analyzer (MCA) obtained from Eurysis Measures, France and Aptec analysing software was used for γ-ray spectroscopic measurements. All the nuclear data used were taken from the publications Table of Isotopes,42 Gamma Ray Catalogue43 and Nuclear Wallet Card.44 A well type NaI(Tl) scintillation counter of Electronic Enterprises Pvt Ltd, Hyderabad, India was used to measure the activity of chromatography paper strip during radiochemical purity assay. Distribution ratios (D) of Pt and Au at different pH were determined following standard radiometric procedures. Nanalysis' NMReady-60e all-in-one benchtop NMR spectrometer with an operation frequency of 60 MHz (1.4 T) was used to record NMR spectra of ethyl acetate.Chemical processing of the irradiated target was carried out in a leak tight isotope processing facility of the dimension 1.8 × 1.8 × 0.9 m3 with 100 mm lead shielding wall all-around and equipped with remote handling tongs, lead viewing windows and other associated accessories.
Neutron irradiation of Pt target
The irradiation container (Fig. 1) essentially consists of a 1S aluminum cylinder of 22 mm (ϕ) × 44.5 mm (h) and a lid. About 100 mg of platinum sponge was accurately weighed, encapsulated in a quartz ampoule, wrapped in aluminum foil, placed in an irradiation container, sealed by cold-pressure-weld and irradiated in the Dhruva research reactor of our institute at a flux of 8 × 1013 n cm−2 s−1 for 7 days. | ||
| Fig. 1 Irradiation container for neutron irradiation (a) schematic drawing (b) photograph. | ||
Determination of distribution ratios of Au3+ and Pt4+ ions in ethyl acetate
With a view to arrive at the optimum separation conditions, distribution ratios (D) of Au3+ and Pt4+ ions in ethyl acetate were determined at different HCl concentration following radiometric technique using 199Au and 197Pt radiotracers. In this procedure, 1 mL of aqueous phase containing varying concentrations of HCl (1–10 M) spiked with either 199Au or 197Pt were equilibrated with equal volume of ethyl acetate in stoppered glass tubes in a thermostated water bath (25 ± 0.1 °C) for 25 min.The two phases were separated, then centrifuged, and 0.1 mL aliquots were taken for radio metric analysis by gamma counting using a well type NaI(Tl) scintillation counter. The distribution ratio (D) was calculated using the expression:

Radiochemical processing and separation of 199Au
The glass ware set up used for the processing of irradiated target is shown in Fig. 2. The irradiated can containing platinum metal target was cut open using a semi-automatic irradiation container opening unit, quartz ampoule containing the irradiated target was cut using a mechanized device, and the radioactive contents were transferred into a 3-necked round bottom flask. 4 mL aqua regia was introduced into the round bottom flask and the target was dissolved by gentle warming to ensure complete dissolution. | ||
| Fig. 2 Experimental set up for production of 199Au. | ||
The solution obtained was evaporated to incipient dryness and then the residue was treated with 5 mL of conc. HCl and heated till evolution of brown fumes ceases. The procedure was repeated thrice to ensure complete removal of HNO3 and the residue left was reconstituted with 10 mL of 5 M HCl. A measured aliquot from this stock was withdrawn and subjected to high resolution gamma ray spectrometry to assess both total 199Au activity produced, and co-produced radionuclide impurities.
The solution was then transferred to a 50 mL separating funnel connected in series with the dissolution flask by applying vacuum. 10 mL ethyl acetate was introduced into the main separating funnel through a side neck. Dry filtered compressed gas was passed through a glass tube from the top of the separating funnel to agitate the solution mixture and to provide a shaking action. Extraction was continued (three times) until nearly all 199Au was extracted into organic phase.
The organic phases (ethyl acetate) were pooled together and heated to remove ethyl acetate by distillation, which was collected and disposed off as radioactive waste. The residue in the flask was treated with 5 mL concentrated HNO3 to destroy any organic matter and evaporated to incipient dryness, reconstituted in 5 mL of 0.1 M HCl gently heated to leach 199Au activity and subsequently withdrawn from the processing flask through a sterile PVC tube with leur ends under suction, passed through a 0.22 μ millipore filter and collected in a sterile vial.
Quality control of 199Au
For radioactivity assay in ionization chamber, the sample in a sealed glass vial was remotely placed at the centre of sensitive volume of ionization chamber and ionization current produced in sensitive zone of instrument was recorded and a conversion factor for 199Au was applied to obtain the value of radioactivity in terms of MBq. The ion current ‘I’ was measured and the radioactivity ‘A’ was calculated by the following relation:

Energy as well as efficiency calibration of the HPGe detector were carried out using a 152Eu reference source prior to the recording of gamma ray spectra of sample. Appropriately diluted aliquots of the processed H[199AuCl4] solution were measured for 1000 seconds. The activity of 199Au in the aliquot was ascertained utilizing the 158.4 keV (36.9%) and 208.2 keV (8.4%) photopeak of 199Au. The absolute activity of the 199Au was calculated from the measured count rate and dividing it by the average efficiency.
Results
Target
While platinum metal remain as the obvious choice for target owing to high melting point, good thermal conductivity and stability under neutron irradiation in nuclear reactors, a holistic consideration should be given whether to use natural Pt or isotopically enriched 198Pt.A distinct advantage of using isotopically enriched 198Pt (∼100%) target is the possibility to produce 199Au with very high radionuclidic purity and offer the prospect of augmenting the yield without significantly altering the radiochemical processing set up. When compared with natural Pt target, use of isotopically enriched 198Pt requires smaller amount of target for neutron irradiation to produce desired amounts of the activity, reduces radiation dose burden during radiochemical processing, generates minor amount of radioactive waste and offers the scope of using low flux reactor. Although the prospect of using isotopically enriched 198Pt is relatively more appealing, the need to recover the expensive target in micro- to milli-gram level for recycling is challenging. In view of this consideration, assessing the potential of natural Pt as target is not only an interesting proposition, but deemed worthy of consideration. In this premise, it is imperative to assess the consequence of using natural Pt.
When natural Pt undergoes irradiation in a reactor, it is crucial to consider all the radionuclides produced by thermal neutrons along with their thermal neutron absorption cross section.45 Table 1 depicts the nuclear reactions taking place when natural Pt undergoes irradiation with thermal neutrons in a nuclear reactor. It is seen from the Table 1 that among all the possible radionuclides formed by natural Pt neutron irradiation, 191Pt, 193Pt, 193mPt, 195mPt, 197Pt and 197mPt are the most prominent. Concomitant production of these radionuclides not only complicate the irradiated target handling owing to augmentation of radiation dose, but also generate radioactive waste. Production of Pt radionuclides other than 199Pt during neutron irradiation, are of least concern as they are unlikely to reduce the specific activity of 199Au because they remain with cold Pt during chemical separation. In view of these considerations, prospect of using natural Pt with some acceptable restrictions is not only an interesting prospect, but deemed worthy of consideration and motivated us to pursue.
| Stable isotope | Isotopic abundance (%) | Nuclear reaction | Reaction cross section (b) | Half life |
|---|---|---|---|---|
| 190Pt | 0.012 | 190Pt(n,γ)191Pt | 152 | 2.83 d |
| 192Pt | 0.782 | 192Pt(n,γ)193Pt | 10 | 50 year |
| 192Pt(n,γ)193mPt | 2.2 | 4.33 d | ||
| 194Pt | 32.86 | 194Pt(n,γ)195Pt | 1.44 | Stable |
| 194Pt(n,γ)195mPt | 0.036 | 4.01 d | ||
| 195Pt | 33.78 | 195Pt(n,γ)196Pt | 27.5 | Stable |
| 196Pt | 25.21 | 196Pt(n,γ)197Pt | 0.58 | 19.89 h |
| 196Pt(n,γ)197mPt | 0.044 | 95.41 min | ||
| 198Pt | 7.36 | 198Pt(n,γ)199Pt | 3.61 | 30.8 min |
| 198Pt(n,γ)199mPt | 0.35 | 13.6 s |
Natural Pt not only co-produces 191Pt, 193Pt, 193mPt, 195mPt, 197Pt and 197mPt due to competing activation reactions but also other radionuclide impurities such as 198Au, 192Ir and 194Ir as a result of neutron activation of some of their decay products. Radionuclides that are likely to be produced during the neutron irradiation of natural Pt are shown in Table 2.
| Radionuclide | Half life | Gamma ray energy | Method of formation of radionuclides | |||
|---|---|---|---|---|---|---|
| keV | % Iγ | Nuclear reaction | % natural abundance | Reaction cross section σth (barns) | ||
| 199Au | 3.139 d | 158.4 | 36.9 |  |
7.36 | 3.61 |
| 208.2 | 8.38 | |||||
| 198Au | 2.695 d | 411.8 | 95.5 |  |
25.21 | 0.58 |
| 98.65 | ||||||
| 192Ir | 73.829 d | 295.9 | 28.7 |  |
0.012 | 152 |
| 308.4 | 29.7 | |||||
| 316.5 | 82.9 | 954 | ||||
| 468.1 | 48.1 | |||||
| 194Ir | 19.28 h | 293.5 | 2.54 |  |
0.782 | 10 |
| 328.4 | 13.0 | 111 | ||||
| 191Pt | 2.83 d | 538.9 | 13.7 | 19078Pt(n,γ)19178Pt | 0.012 | 152 |
| 409.4 | 8 | |||||
| 193mPt | 4.33 d | 66.8 | 7.38 | 19278Pt(n,γ)193m78Pt | 0.782 | 2.2 |
| 75.7 | 2.53 | |||||
| 135.5 | 0.11 | |||||
| 195mPt | 4.01 d | 98.9 | 11.4 | 19478Pt(n,γ)195m78Pt | 32.86 | 0.036 |
| 129.7 | 2.83 | |||||
| 197Pt | 19.891 h | 77.3 | 17.2 | 19678Pt(n,γ)19778Pt | 25.21 | 0.58 |
| 191.4 | 3.68 | |||||
| 197mPt | 95.41 min | 66.8 | 23.8 | 19678Pt(n,γ)197m78Pt | 25.21 | 0.044 |
| 364.5 | 11.2 | |||||
Optimization of irradiation parameters
With an aim to produce 199Au of required quality and quantity, the irradiation time and neutron flux were judiciously optimized.The 199Au activity produced at the end of neutron irradiation as a function of irradiation time for different thermal neutron fluxes has been calculated and the results are shown in Fig. 3. There appears to be enticing interest to consider irradiation of Pt at higher thermal neutron flux and shorter irradiation period as it will suffice to obtain requisite 199Au yield.
 | ||
| Fig. 3 Gold-199 (199Au) activity produced (MBq) per mg platinum target as a function of the irradiation time at different thermal neutron flux of research reactor. | ||
Owing to the inherent limitation on the availability of limited number of irradiation position at highest neutron flux 1.8 × 1014 n cm−2 s−1 in a multi-purpose research reactor and their exclusive utilization for routine production of 99Mo and 177Lu etc., it was not possible to perform irradiation. Consequently, our subsequent efforts were directed towards exploring the possibility to produce 199Au at a neutron flux of 8 × 1013 n cm−2 s−1. Typical calculated yields of 199Au from natural and enriched targets for different durations of irradiation in Dhruva reactor at a neutron flux of 8 × 1013 n cm−2 s−1 are shown in Fig. 4.
 | ||
| Fig. 4 Typical calculated yields of 199Au produced from natural and enriched target of Pt for different irradiation time at a thermal neutron flux of 8 × 1013 n cm−2 s−1. | ||
As expected, the yield of 199Au in natural target increases marginally with increasing irradiation time. In the case of the 95% enriched 198Pt target, increase in irradiation time not only increase the yield significantly, but also significantly higher than those obtained from natural Pt targets under similar irradiation conditions.
The irradiation time of 7 d was deemed optimum from the prospective of attaining adequate radioactivity yield of 199Au and inhibits built up of longer isotopes of Pt (e.g. 193Pt) and radioactive impurities. In order to avail GBq amounts of 199Au per batch, irradiation of 100 mg nat. Pt target is necessary. It is pertinent to point out that 2.7 mg 95% enriched target of Pt (I.E 198Pt) under similar conditions of irradiation would be able to provide the same quantity of 199Au.
Target processing
In view of the need to dissolve the neutron irradiated platinum quantitatively for subsequent radiochemical separation, the scope of using aqua-regia seemed attractive.The initial chemical reactions of Pt with aqua-regia, led to the formation of mixture of chloroplatinous acid (H2PtCl4) and nitrosoplatinic chloride [(NO)2PtCl4] in solution.
| 2Pt(s) + 2HNO3(aq) + 8HCl(aq) → (NO)2PtCl4(s) + H2PtCl4(aq) + 4H2O(l) |
When this irradiated Pt was treated with aqua-regia, it steadily dissolves, but left a solid residue due to formation of nitrosoplatinic chloride, which subsequently dissolved with time due to the formation of chloroplatinous acid.
| (NO)2PtCl4(s) + 2HCl(aq) → H2PtCl4(aq) + 2NOCl(g) |
The chloroplatinous acid (H2PtCl4) was oxidized to chloroplatinic acid with chlorine while heating.
| H2PtCl4(aq) + Cl2(g) → H2PtCl6(aq) |
The overall equation of Pt dissolution can therefore be written as
| 2Pt(s) + 4HNO3(aq) + 12HCl(aq) + Cl2(g) → 2H2PtCl6(aq) + 6H2O(l) + O2(g) + 4NOCl(g) |
The corresponding equation for Au is
| Au(s) + 3HNO3(aq) + 4HCl(aq) → H[AuCl4](aq) + NO2(g) + 3H2O(l) |
Excess of residual nitric acid was removed by repeated treatment with conc. HCl and heating.
The gamma spectrum of the solution as shown in Fig. 5 shows gamma peaks pertaining to only Pt and Au radionuclides, which indicated that the target used for 199Au production was free from other metallic impurities. The gamma spectrum further revealed that radionuclides formed apart from 199Au were 191Pt, 193mPt, 195mPt and 197Pt. It was further observed that 192Pt, 197mPt, 198Au, 192Ir and 194Ir which are expected to be formed during neutron irradiation were below detectable levels. Their absence is primarily attributed due to combination of multiple factors such as low abundance of corresponding target nuclide, low neutron absorption cross section, short irradiation time and long half-life of the product radionuclide etc.
 | ||
| Fig. 5 Gamma ray spectrum of the feed solution before radiochemical separation. | ||
Following the aforementioned procedure, several batches of 199Au were produced by irradiating 20 mg nat. Pt target for 7 d at various neutron flux in Dhruva research reactor, activity of 199Au produced were measured following gamma spectrometric technique and the result obtained are depicted in Table 3. As expected, the activity produced increases with increasing the neutron flux. Activity measurement from gamma spectrometric technique revealed that in a typical batch, about 290–936 MBq (7.8–25.3 mCi) of 199Au could be produced by irradiating 20 mg platinum target.
| Batcha | Thermal neutron flux n cm−2 s−1 | Activity producedb MBq (mCi) |
|---|---|---|
| a Average of five batches.b Activity corrected to EOI-end of irradiation. | ||
| 1 | 2.3 ± 0.2 × 1013 | 288 ± 7 (7.8 ± 0.2) |
| 2 | 3.4 ± 0.2 × 1013 | 407 ± 74 (11 ± 2) |
| 3 | 4.2 ± 0.3 × 1013 | 540 ± 7 (14.6 ± 0.2) |
| 4 | 5.6 ± 0.2 × 1013 | 740 ± 111 (20 ± 3) |
| 5 | 7.5 ± 0.1 × 1013 | 936 ± 11 (25.3 ± 0.3) |
Radiochemical separation of 199Au from neutron irradiated Pt
Following the aforementioned procedure, D values of Au and Pt ions were determined at different HCl concentrations and the results are summarized in Table 4.| HCl concentration (M) | D | |
|---|---|---|
| 199Au | 197Pt | |
| 1 | 5.3 ± 0.1 | 0.06 ± 0.01 |
| 2 | 7.0 ± 0.1 | 0.06 ± 0.01 |
| 3 | 10.3 ± 0.2 | 0.07 ± 0.02 |
| 4 | 11.6 ± 0.4 | 0.05 ± 0.01 |
| 5 | 20.0 ± 0.5 | 0.07 ± 0.02 |
| 6 | 7.7 ± 0.6 | 0.08 ± 0.01 |
| 7 | 6.1 ± 0.8 | 0.01 ± 0.01 |
| 8 | 6.1 ± 0.1 | 0.08 ± 0.02 |
| 9 | 4.2 ± 0.3 | 0.07 ± 0.01 |
| 10 | 1.2 ± 0.1 | 0.11 ± 0.03 |
A careful scrutiny of the D values of Au indicates that it increases with increasing acidity and attains a maximum at 5 M HCl and thereafter decreases. Pt has negligible D values in ethyl acetate at all the acidity of HCl studied. The most striking feature of Au extraction in ethyl acetate is its extremely high D values compared with that of Pt in all acidity studied. From the perspective of isolation 199Au, the excellent distribution ratio (D) values for 199Au over Pt at 5 M HCl is particularly heartening and conducive for the selective recovery of 199Au. This assumption has been simplified and assiduously exploited in our work.
While the distribution ratio values are beneficial to achieve clean separation, it is obligatory to evaluate the behavior of ethyl acetate in the presence of intense radiation environment with the radiolytic products generated as a result of radioactive isotopes of Pt and 199Au. In view of these considerations, validation of the separation procedure was carried out using 20 mg of irradiated Pt target following the procedure in the Experimental section.
While the use of single extraction of 199Au had tangible benefits, but following multiple extractions (3 times) was considered a trustworthy proposition. Experimentally, it was seen that it was possible to extract >75% of 199Au in a single extraction, where as >95% yield was achievable in 3 extractions. A reproducible separation efficiency of 94–97% was found to be achieved in 3 extractions. With an aim to verify the efficacy of the 3 step extraction procedure, gamma spectrum of an aliquot withdrawn from the aqueous fraction was taken as shown in Fig. 6. The gamma spectrum obtained shows photo-peaks pertaining to Pt radionuclides only. Absence of photo-peak corresponding to 199Au in the aqueous phase confirms quantitative transfer of 199Au into the organic phase.
 | ||
| Fig. 6 Gamma ray spectrum of the feed solution after radiochemical separation of 199Au. | ||
Having successfully completed the feasibility demonstration studies, scaling-up of the optimized radiochemical separation procedure was then undertaken by gradual increase of the weight of the target and it was observed that with increasing the batch size, no major problems in term of reproducibility of purity or yield of 199Au were encountered during the production process.
The aforementioned developed radiochemical separation process was repeated in 5 different batches using 100 mg of neutron irradiated Pt target irradiated for 7 d at 8 × 1013 n cm−2 s−1 and results obtained is shown in Table 5.
| Batch no. | Activity before chemical separation (GBq) | Activity recovered (GBq) | Recovery yield (%) |
|---|---|---|---|
| 1 | 5.55 ± 0.25 | 5.16 ± 0.21 | 92.97 |
| 2 | 5.72 ± 0.22 | 5.49 ± 0.26 | 95.97 |
| 3 | 5.63 ± 0.28 | 5.41 ± 0.21 | 96.09 |
| 4 | 4.81 ± 0.23 | 4.63 ± 0.25 | 96.25 |
| 5 | 5.75 ± 0.19 | 5.56 ± 0.27 | 96.69 |
Experimental results demonstrated that it was possible to produce about 5.6 GBq 199Au per batch from 100 mg of irradiated Pt using the optimized procedure.
Quality control of 199Au
 | ||
| Fig. 7 Gamma spectra of separated 199Au–HAuCl4 solution. | ||
The gamma spectrum of the decayed sample did not show any photo peaks thereby confirmed the absence of any long lived extraneous radionuclide in the product.
 | ||
| Fig. 8 Paper chromatographic pattern of 199Au–HAuCl4 using saline in 0.02 M HCl as the eluting solvent. | ||
Gold-199 (199Au) product specification
Chemical form the product obtained was HAuCl4 in 0.1 M HCl, appearance was a clear solution, and the radioactive concentration was 370–740 MBq (10–20 mCi) per mL. Radionuclidic purity attained was >99.9. Radiochemical purity of the 199Au obtained was greater than 99% in all the lots analyzed.Radioactive waste management
The yields of various coproduced Pt radionuclides that constitute the major radionuclide impurities are given in Table 6. Although, concomitant generation of radioactive waste containing medium energy gamma emitting radionuclides is a matter of concern from the prospective of waste management point of view, but surmountable owing to their relatively short half-lives. Aqueous radioactive waste were collected in a labeled container kept in the shielded enclosure of radiochemical processing facility and allowed to decay for 30 days and disposed.| Quantity of Pt irradiated (mg) | Radio nuclide | Activity at EOI MBq (mCi) | Activity per mg Pt at EOI MBq (mCi) |
|---|---|---|---|
| 20 | 199Au | 288 ± 7 (7.8 ± 0.2) | 14.4 (0.39) |
| 191Pt | 11.8 ± 0.4 (0.32 ± 0.01) | 0.59 (0.016) | |
| 195mPt | 14.4 ± 0.7 (0.39 ± 0.02) | 0.72 (0.019) | |
| 197Pt | 331.5 ± 19.9 (8.96 ± 0.54) | 16.6 (0.448) | |
| 100 | 199Au | 1554 ± 185 (42 ± 5) | 15.5 (0.42) |
| 191Pt | 66.6 ± 7.4 (1.8 ± 0.2) | 0.66 (0.018) | |
| 195mPt | 77.7 ± 7.4 (2.1 ± 0.2) | 0.77 (0.021) | |
| 197Pt | 1676.1 ± 88.8 (45.3 ± 2.4) | 16.7 (0.453) |
Discussion
While both the reactor and accelerator production 199Au routes are pursued, either of them is not pitted against each other, but instead, capable of providing clinical grade NCA 199Au. Each institution needs to assess options, weigh pros and cons and select the one based on the available technical and economic resources to suit its needs. Our pursuit to develop a method for the routine production of 199Au is driven due to the availability of operating research reactor of neutron flux of the order 1014 n cm−1 s−1 as well as expertise and shielded radiochemical facilities to undertake chemical processing of neutron irradiated target in our institution.The prospect of using a volatile extractant for the selective extraction of 199Au seemed sagacious as it provides the possibility of recovering 199Au by evaporation and subsequent leaching from glass vessel.
Ethyl acetate is susceptible to radiolytic degradation, especially when GBq activity of 199Au are processed. NMR spectra of ethyl acetate before and after radiochemical separation were recorded and depicted in Fig. 9. As seen from the NMR spectra, there is significant change in the chemical composition of ethyl acetate on exposure to GBq levels of radioactivity. In order to circumvent the radiolytic degradation of ethyl acetate, the extraction is repeated three times with fresh ethyl acetate each time.
 | ||
| Fig. 9 NMR spectrum of ethyl acetate (a) before radiochemical separation (b) after radiochemical separation. | ||
The flow sheet of the radiochemical separation process is depicted in Fig. 10.
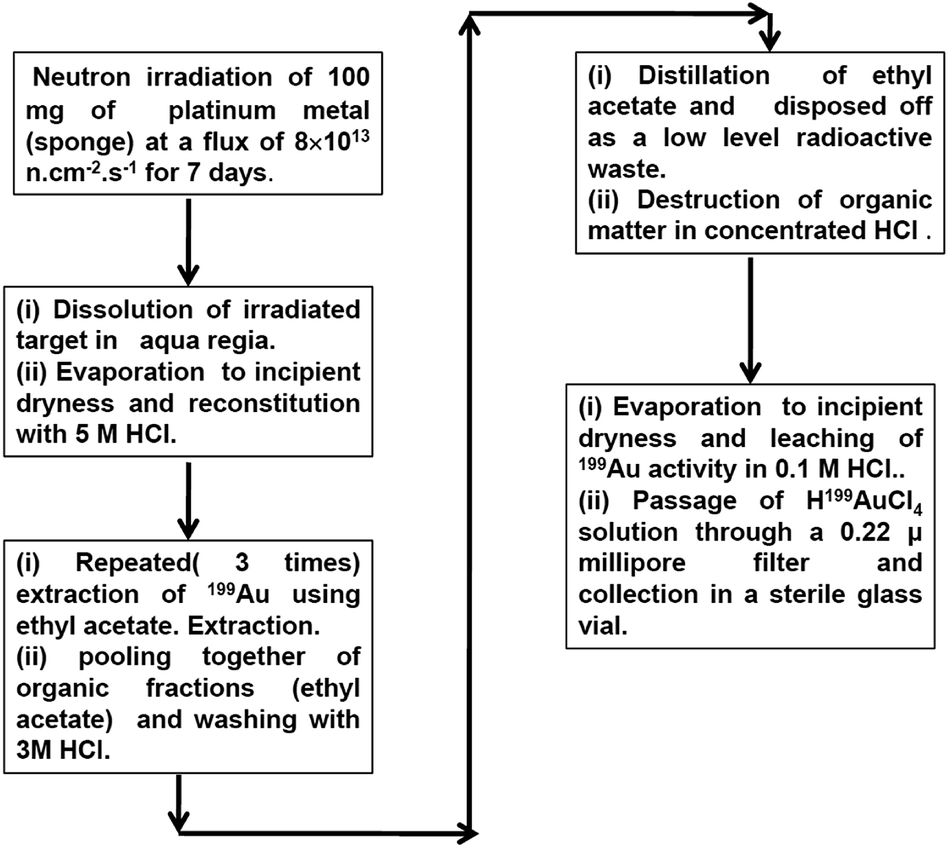 | ||
| Fig. 10 Flow sheet of the radiochemical separation process to isolate 199Au from neutron irradiated Pt. | ||
Given the simplicity of producing 199Au by reactor activation of natural Pt targets and ease of adaptation of radiochemical separation strategy, the reported procedure would be of considerable value for accessing NCA 199Au.
Conclusions
The objective of developing a facile and robust procedure to undertake routine production of NCA 199Au using natural Pt target has been successfully achieved. The developed technology is sufficed to provide 5.6 GBq (∼150 mCi) of NCA 199Au per batch. Using this procedure it is possible to avail 199Au in >95% yields with acceptable radionuclidic and radiochemical purity. The method can be upscale to the desired level if situations arise. It is envisaged that the reported production procedure would serve in good stead for ensuring local availability of 199Au. The ubiquitous cost effective availability of 199Au would facilitate more research on new radiopharmaceuticals with 199Au for therapy.Conflict of interest
The authors declared that they have no financial or other conflict of interest relevant to the subject of this article.Acknowledgements
Research at the Bhabha Atomic Research Centre is part of the ongoing activities of the Department of Atomic Energy, India and is fully supported by government funding.Notes and references
- C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2013, 113(2), 858–883 CrossRef CAS PubMed.
- M. Neves, A. Kling and A. Oliveira, J. Radioanal. Nucl. Chem., 2005, 266, 377–384 CrossRef CAS.
- C. Cutler, P. Kan, N. Chanda, S. Jurisson, L. D. Watkinson, J. R. Lever, J. C. Smith, K. V. Katti, R. Kannan and K. Katti, Trans. Am. Nucl. Soc., 2010, 103, 1123–1124 Search PubMed.
- P. Anderson, A. T. Vaughan and N. R. Varley, Nucl. Med. Biol., 1988, 15(3), 293–297 CAS.
- J. L. Humm, J. Nucl. Med., 1986, 27(9), 1490–1497 CAS.
- J. F. Hainfeld, Nature, 1988, 333, 281–282 CrossRef CAS PubMed.
- J. F. Hainfeld, Science, 1987, 236, 450–453 CAS.
- J. F. Hainseld, C. I. Foley, S. C. Srivastava, L. F. Mausner, N. I. Feng, G. E. Meinken and Z. Steplewski, Nucl. Med. Biol., 1990, 17, 287–294 Search PubMed.
- K. L. Kolsky and L. F. Mausner, Appl. Radiat. Isot., 1993, 44(3), 553–560 CrossRef CAS PubMed.
- Y. Zhao, B. Pang, H. Luehmann, L. Detering, X. Yang, D. Sultan, S. Harpstrite, V. Sharma, C. S. Cutler, Y. Xia and Y. Liu, Adv. Healthcare Mater., 2016, 5(8), 928–935 CrossRef CAS PubMed.
- Y. Fazaeli, O. Akhavan, R. Rahighi, M. R. Aboudzadeh, E. Karimi and H. Afarideh, Mater. Sci. Eng., C, 2014, 45, 196–204 CrossRef CAS PubMed.
- N. Chanda, P. Kan, L. D. Watkinson, R. Shukla, A. Zambre, T. L. Carmack, H. Engelbrecht, J. R. Lever, K. Katti, G. M. Fent, S. W. Casteel, C. J. Smith, W. H. Miller, S. Jurisson, E. Boote, J. D. Robertson, C. Cutler, M. Dobrovolskaia, R. Kannan and K. V. Katti, Nanomedicine, 2010, 6, 201–209 CAS.
- R. Shukla, N. Chanda, A. Zambre, A. Upendran, K. Katti, R. R. Kulkarni, S. K. Nune, S. W. Casteel, C. J. Smith, J. Vimal, E. Boote, J. D. Robertson, P. Kan, H. Engelbrecht, L. D. Watkinson, V. Carmack, J. R. Lever, C. S. Cutler, C. Caldwell, R. Kannan and K. V. Katti, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12426–12431 CrossRef CAS PubMed.
- D. Suresh, A. Zambre, N. Chanda, T. J. Hoffman, C. J. Smith, J. D. Robertson and R. Kannan, Bioconjugate Chem., 2014, 25, 1565–1579 CrossRef CAS PubMed.
- R. Kannan, A. Zambre, N. Chanda, R. Kulkarni, R. Shukla, K. Katti, A. Upendran, C. Cutler, E. Boote and K. V. Katti, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 42–51 CrossRef CAS PubMed.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- K. C. L. Black, Y. Wang, H. P. Luehmann, X. Cai, W. Xing, B. Pang, Y. Zhao, C. S. Cutler, L. V. Wang, Y. Liu and Y. Xia, ACS Nano, 2014, 8, 4385–4394 CrossRef CAS PubMed.
- R. Kannan, V. Rahing, C. Cutler, R. Pandrapragada, K. K. Katti, V. Kattumuri, J. D. Robertson, S. J. Casteel, S. Jurisson, C. Smith, E. Boote and K. V. Katti, J. Am. Chem. Soc., 2006, 128, 11342–11343 CrossRef CAS PubMed.
- K. V. Katti, R. Kannan, K. Katti, V. Kattumori, R. Pandrapraganda, V. Rahing, C. Cutler, E. J. Boote, S. W. Casteel, C. J. Smith, J. D. Robertson and S. S. Jurrison, Czech. J. Phys., 2006, 56, D23–D34 CrossRef CAS.
- A. Mushtaq, Open Access Library Journal, 2014, 1, 1–9 Search PubMed.
- F. F. Knapp (Russ) Jr, S. Mirzadeh and A. l. Beets, J. Radioanal. Nucl. Chem., 1996, 205, 93–100 CrossRef.
- M. Neves, A. Kling and R. M. Lambrecht, Appl. Radiat. Isot., 2002, 57, 657–664 CrossRef CAS PubMed.
- F. F. Knapp (Russ) and A. Dash, Reactor-Produced Therapeutic Radionuclides, in Radiopharmaceuticals for Therapy, Springer, India, 2015, pp. 71–113 Search PubMed.
- A. R. Ketring, C. S. Cutler and G. J. Ehrhardt, Trans. Am. Nucl. Soc., 2008, 98, 871 Search PubMed.
- P. Kan, H. K. Engelbrecht, G. J. Ehrhardt, C. Cutler and S. S. Jurisson, Proc of the 236th ACS National Meeting, Philadelphia, PA, August 2008 Search PubMed.
- M. Bonardi, F. Groppi, C. Birattari and D. Arginelli, Ann. Chim., 2002, 92, 795–813 CAS.
- M. Akhter, A. Mushtaq, H. M. A. Karim and M. A. Khan, Radiochim. Acta, 1994, 64, 137–138 Search PubMed.
- F. Tárkányi, S. Takács, F. Ditrói, A. Hermanne, Y. N. Shubin and A. I. Dityuk, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 226, 490–498 CrossRef.
- F. Tarkanyi, A. Hermanne, F. Ditroi, S. Takacs, R. Adam-Rebeles and A. V. Ignatyuk, Nucl. Instrum. Methods Phys. Res., Sect. B, 2015, 362, 116–132 CrossRef CAS.
- F. Tarkányi, A. Hermanne, S. Takács, Y. N. Shubin and A. I. Dityuk, Radiochim. Acta, 2004, 92(4–6), 223–228 CrossRef.
- M. U. Khandaker, H. Haba, M. Murakami, N. Otuka and H. A. Kassim, Nucl. Instrum. Methods Phys. Res., Sect. B, 2015, 362, 151–162 CrossRef CAS.
- M. U. Khandaker, H. Haba and H. A. Kassim, AIP Conf. Proc., 2016, 1704, 030008 CrossRef.
- F. Ditroi, F. Tárkányi, J. Csikai, M. S. Uddin, M. Hagiwara, M. Baba, Y. N. Shubin and S. F. Kovalev, Nucl. Instrum. Methods Phys. Res., Sect. B, 2006, 243, 20–27 CrossRef CAS.
- E. McMillan, M. Kamen and S. Ruben, Phys. Rev., 1937, 52, 375–377 CrossRef CAS.
- S. M. Hasany, I. Hanif and I. H. Qureshi, Int. J. Appl. Radiat. Isot., 1978, 29, 145–149 CrossRef CAS.
- Li. Chunsheng, C. Chai, X. Yang, X. Hou and X. Mao, Talanta, 1997, 44, 1313–1317 CrossRef.
- L. F. Mausner, K. L. Kolsky, V. Joshi and S. C. Srivastava, Appl. Radiat. Isot., 1998, 49(4), 285–294 CrossRef CAS PubMed.
- N. R. Das, S. Banerjee, K. Chatterjee and S. Lahiri, Appl. Radiat. Isot., 1999, 50, 643–647 CrossRef CAS.
- N. R. Das, S. Banerjee, K. Chatterjee and S. Lahiri, Radiochim. Acta, 1998, 83, 39–42 CrossRef CAS.
- J. Korkisch and H. Klakl, Talanta, 1968, 15, 339–346 CrossRef CAS PubMed.
- P. Benes and J. Smetana, Radiochim. Acta, 1966, 6, 196–201 CrossRef.
- Table of isotopes, ed. R. Firestone and V. S. Shirley. John Wiley & Sons Inc., New York, 8th edn, 1996 Search PubMed.
- U. Reus, W. Westmeier and I. Warnecke, At. Data Nucl. Data Tables, 1983, 193–406 CrossRef CAS.
- J. K. Tuli, Nuclear Wallet Cards, National Nuclear Data Center, USA, 8th edn, 2011 Search PubMed.
- IAEA Nuclear data Cross section, International Atomic Energy Agency (IAEA), Vienna, Austria, accessed on 24 May 2016, available at: https://www-nds.iaea.org/relnsd/NdsEnsdf/neutroncs.html Search PubMed.
- S. Chakraborty, K. V. Vimalnath, S. P. Lohar, P. Shetty, A. Dash and J. Radioalnal, Nucl. Chem., 2014, 302, 233–243 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |