
Facile and controllable synthesis of triplex Au@Ag–Pt@infinite coordination polymer core–shell nanoparticles for highly efficient immobilization of enzymes and enhanced electrochemical biosensing activity†
Lihua Wang‡
ab,
Yi Zeng‡a,
Aiguo Shen*a,
Yingchun Fu c,
Lingwen Zengb and
Jiming Hua
c,
Lingwen Zengb and
Jiming Hua
aKey Laboratory of Analytical Chemistry for Biology and Medicine, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, 430072 Wuhan, P. R. China. E-mail: agshen@whu.edu.cn; Fax: +86 27 68752136
bInstitute of Environmental and Food Safety, Wuhan Academy of Agricultural Science and Technology, Wuhan, 430207, P. R. China
cLaboratory of Biosensing and Biological Modelling, School of Biosystems Engineering and Food Science, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China
First published on 25th August 2016
Abstract
We present a simple and controllable strategy to synthesize triplex noble metal nanoparticle (NP)-mixed infinite coordination polymer (ICP) core–shell heterostructures (Au@Ag–Pt@ICPs) that are capable of acting as an efficient host matrix for in situ immobilization of enzymes for highly efficient electrochemical biosensing. In this synthesis, both H2PtCl6 and nanoscale metallic Ag (Au@Ag NP) are demonstrated to simultaneously undergo a coordination reaction with an organic ligand, 2,5-dimercapto-1,3,4-thiadiazole, based on a systematic and comprehensive analysis. Besides, the size of the ICP shells can be efficiently tuned by modulation of the amount of DMcT, H2PtCl6 and the size ratio of Au@Ag NPs. Using glucose oxidase as a model enzyme, the Au@Ag–Pt@ICPs based biosensors achieve enhanced electrochemical biosensing performances (e.g., sensitivity, stability and else) in comparison with the pure ICP NP-based biosensors due to the synergistic effects of ICPs and noble metal NPs. Under optimized conditions, they offer a much wider linear range (from 0.5 μM to 3.33 mM), an extremely low detection limit (60 nM, S/N = 3), a very high sensitivity (82.1 μA mM−1 cm−2) and a very high biological affinity (the apparent Michaelis–Menten constant was estimated to be 0.28 mM) as well as good thermal stability and long-term stability. The presented experimental platform/strategy may be extended to the preparation of many other ICP containing bio/nano hybrid materials.
1. Introduction
Biosensing of target analytes is regarded as one of the most important issues in several academic and industrial fields, such as bioanalysis, environmental monitoring, and food safety.1 To date, there are a few potential sensing modalities, including optical,2 acoustic,3 and transdermal technologies,4 being explored. Among these methods, electrochemical sensors are recognized universally as the most convenient and effective tools for target analysis owing to their attractive features such as pre-eminent sensitivity, time efficiency, simple instrumentation, easy operation, and low production cost.5 A long-term challenge of electrochemical biosensing is how to effectively immobilize the bio-recognition molecules (e.g., enzymes) in constructing high-performance biosensors, since the loading/activity of the immobilized bio-recognition molecules can be largely affected.1,6 Until now, there have been several common approaches for the immobilization of enzymes such as physical adsorption,7 entrapment,8 covalent and cross-linking.9 As one of the most commonly immobilization approaches for enzymes, the entrapment method has been explored widely and many excellent biosensors have been developed to meet a range of different requirements.10–13 Although progress has been made, improving the enzymes' loading capacity/activity and the biosensor's storage stability is still challenging. To resolve these problems, more and more researchers are focusing on developing novel nanomaterials as host matrixes for the immobilization of enzymes, which have been demonstrated to contain lots of unusual advantages in electro-analysis.14–16Infinite coordination polymer nanoparticles (ICP NPs), which are constructed by coordination polymerization between metal ions or metal ion clusters and polydentate bridging ligands, are a typical example. Among a large number of candidate chemosensory materials, water stable ICPs have become particularly attractive in biosensor development for their significant advantages: (1) the simple and mild synthetic procedures usually consist of a one-pot reaction between metal salts and bifunctional ligand precursors at room temperature; (2) the three-dimensional (3D) and highly porous structures as well as excellent structural tailorability allow them to effectively encapsulate guest materials, especially water-soluble molecules (such as dyes, proteins, and so on) and metal NPs. So far, several kinds of water stable and adaptive materials for biosensing have been developed. For example, Mao et al. successfully fabricated an excellent glucose dehydrogenase (GDH)-based biosensor by including all biosensing elements such as an electro-catalyst (methylene green) and GDH during the self-assembly process of NAD+ and Tb3+ ions.17 Similarly, Xie et al. also prepared ICPs based on a ligand, 2,5-dimercapto-1,3,4-thiadiazole (DMcT), and two metallic salts and successfully exploited them as new efficient matrixes to in situ immobilize enzymes for detecting glucose or phenols.1 Despite this research, the direct use of single component ICPs in electrochemistry is limited due to their poor electronic conductivities, low mechanical stabilities and inferior electro-catalytic abilities, which are harmful for the construction of excellent biosensors with high sensitivity and stability. To better address these significant issues, researchers attempted to explore the hetero-structures integrating ICPs with other highly conductive and mechanically durable materials. For example, loading-type ICP hetero-structures, which are prepared through loading the ICP NPs on the surface of other functional materials (e.g., single-walled carbon nanotubes,18 graphene nanosheets,19 etc.), have been demonstrated to exhibit enhanced biosensing properties due to the synergism effect of the functional materials and ICPs. By contrast, research on ICP core–shell heterostructures with functional materials as a core and ICPs as the shell have not been popular to date. In fact, the potential multifunctions of the ICP core–shell heterostructures are attractive as we can rationally utilize the combination of high porosity and high structural tailorability of the ICP shell, as well as the application of the core functional material we choose. For instance, noble metal NPs not only possess excellent conductivity and catalytic activity but also can provide a suitable biocompatible microenvironment for the biomolecules to retain their activity once they are immobilized,20–22 which makes them suitable and popular for the fabrication of electrochemical biosensors.23,24 Predictably, such noble metal NPs@ICPs heterostructures should possess potential application in electrochemical biosensors with a high load/activity of the immobilized biomolecules and enhanced catalytic activity. To our knowledge, such attempts have not yet been reported.
Herein, we present a facile and controllable method to fabricate triplex Au@Ag–Pt@ICPs NPs as new host matrixes for the in situ immobilization of enzymes for electrochemical biosensing, based on the replacement reaction of Au@Ag NPs and H2PtCl6, as well as the coordination reactions of DMcT, extra Ag(0) and H2PtCl6. In this synthesis, the DMcT related aqueous-phase coordination polymerization reactions have been studied by FT-IR, Raman spectroscopy, microscopic techniques, and X-ray photoelectron spectroscopy (XPS) analysis. Besides, the size of the ICPs shell can be efficiently tuned by modulation of the amount of DMcT, H2PtCl6 and the composition of the Au@Ag NPs. Moreover, to demonstrate the high efficiency of Au@Ag–Pt@ICPs NPs as a host matrix of biomolecules, GOx (a model enzyme, which is most popular and widely studied for biosensing) was used. It is found that the Au@Ag–Pt@ICPs NP-based glucose biosensor exhibits a much better performance (very high sensitivity, excellent thermal stability and long-term stability) in comparison to those solely prepared with ICPs and most of the reported analogues.
2. Experimental
2.1 Chemicals
Tetrachloroauric(III) acid and sodium citrate were purchased from Sigma (USA). Silver nitrate was obtained from Beijing Chemical Reagents Company (Beijing, China). Ascorbic acid and chloroplatinic acid were supplied by Sinopharm Chemical Reagent Co., Ltd. 2,5-Dimercapto-1,3,4-thiadiazole (DMcT, ≥98%) was a product of Alfa Aesar. GOx (EC1.1.3.4; type II from Aspergillus niger, activity ≈ 150 kU g−1) was purchased from Sigma. A phosphate buffer solution (PBS, pH 7.0), 0.1 M KH2PO4–Na2HPO4 + 0.1 M K2SO4, was used. All other chemicals were of analytical grade or better quality and used as received. Milli-Q ultrapure water (Millipore, ≥18 MΩ cm) was used throughout.2.2 Preparation of Au@Ag–Pt@ICPs NPs
For the preparation of the Au@Ag–Pt@ICPs core–shell NPs, typically, 80 μL of a 0.038 M H2PtCl6 aqueous solution was introduced into 2 mL of an Au@Ag colloid solution as synthesized in our previous research25 and then vigorously stirred for 30 min. Subsequently, 1 mL of freshly prepared 0.5 mg mL−1 DMcT aqueous solution (ultrasonically dispersed in a PB suspension (0.1 M, pH 7.4)) was added dropwise to the dispersion under continuous stirring. After stirring for about 3 h at room temperature, the product was collected by centrifugation, and resuspended with deionized water three times.2.3 Preparation of the enzyme electrodes
The procedure for the preparation of the Au@Ag–Pt@ICPs–GOx biocomposites is as the same as for the preparation of the Au@Ag–Pt@ICPs NPs, with only 9.0 mg GOx dissolved in the above DMcT aqueous solution. Then, 1 mL of an Au@Ag–Pt@ICPs–GOx aqueous suspension as synthesized was finally concentrated and redispersed in 150 μL water.The bare Pt electrodes were cleaned thoroughly according to a reported procedure with minor modifications.26 Then, 5 μL of the Au@Ag–Pt@ICPs–GOx aqueous suspension was carefully cast onto the Pt electrode and dried at room temperature (Au@Ag–Pt@ICPs–GOx/Pt). Pt electrodes modified by ICPs–GOx (ICPs–GOx/Pt), Au–ICPs–GOx (Au–ICPs–GOx/Pt), Au@Ag@ICPs–GOx (Au@Ag@ICPs–GOx/Pt), Au-pre-synthesized Pt NPs–ICPs–GOx (Au-pre-synthesized Pt NPs–ICPs–GOx/Pt) and Au@Ag-pre-synthesized Pt NPs–ICPs–GOx (Au@Ag-pre-synthesized Pt NPs–ICPs–GOx/Pt) were also prepared for control experiments. Among them, small Pt NPs were pre-synthesized according to previous research.27 The experimental details are given in the ESI.† When not in use, all of the biosensors were stored in 0.1 M PBS at pH 7.0 and 4 °C.
2.4 Amperometric biosensing measurements
Measurements of the prepared enzyme electrodes were carried out at 0.7 V for glucose under solution-stirred conditions in 0.1 M PBS at pH 7.0, and the response current was marked with the change value between the steady-state current after addition of a substrate and the initial background current without the substrate.2.5 Measurements
All electrochemical experiments were conducted on a CHI 660A electrochemical workstation using a conventional three-electrode electrolytic cell. Pt disk electrodes with a 3.0 mm diameter (0.071 cm2 area) served as the working electrodes, a KCl-saturated calomel electrode (SCE) was the reference electrode, and a carbon rod was the counter electrode. All potentials are cited versus SCE. UV-vis studies were performed on a UV spectrometer (Shimadzu, UV-2550). FT-IR spectra were collected on a pressed pellet with KBr in the transmission mode on a Nexus-5700 Fourier transform-infrared spectrophotometer (Nicolet Instrument Co.). Raman experiments were conducted on a confocal microprobe Raman spectroscopy (Jobin Yvon Horiba, France) with a 632.8 nm helium-neon laser at 13.6 mW with a 2 μm spot size. The objective used was an Olympus 50× long working distance lens with an air-cooled charge-coupled device detector. The slit width of the pin hole was set as 200 μm. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Model ESCALAB 250 (Thermo Fisher Scientific) apparatus with an Al Kα X-ray source (1486.6 eV, 15 kV, 150 W) and operated at a pressure greater than 2 × 10−9 Pa. All of the spectra were charge shift corrected with reference to the C1s peak at 284.8 eV. Scanning electron microscope (SEM) images were obtained by Sigma, ZEISS. A JEOL JEM-2100 TEM equipped with an EDX analyzer was used to determine the morphology and composition of products.2.6 Live subject statement
To conduct a determination of glucose in human serum samples, three fresh blood samples were obtained from the Department of Oncology, Zhongnan Hospital of Wuhan University. All experiments were performed in compliance with the laws of People's Republic of China and approved by the Medical Ethics Committee of Zhongnan Hospital, Wuhan University.3. Results and discussion
3.1 Synthesis and characterization of Au@Ag–Pt@ICPs NPs
As illustrated in Fig. 1, Au@Ag NPs were first prepared through the reduction of AgNO3 by ascorbic acid at room temperature. A galvanic replacement reaction was then performed for 30 min with the addition of some volume of H2PtCl6 to an aqueous solution of the synthesized Au@Ag NPs to obtain Au@Ag–Pt nanostructures. During this period, a clear colour change from orange in the Au@Ag NP colloid solution to purple and finally black was observed, which suggest small-sized Pt NPs surrounding single Au@Ag NPs formed. Subsequently, a DMcT aqueous solution was then introduced and simultaneously reacted with the Au@Ag–Pt nanostructures and the extra H2PtCl6 for about 3 hours. Due to the coordination reactions between DMcT, Ag(0) and H2PtCl6, the ICPs would form around the Au@Ag–Pt, and a new core–shell nanostructure was finally produced (Au@Ag–Pt@ICPs). Fig. S1† displays the extinction spectra of the whole reaction system at different stages in this synthetic process. First, there are two peaks at ca. 392 and 486 nm in the spectrum of the original Au@Ag NPs. Our previous work attributed these two peaks to the surface plasmon resonance absorption of the silver shell and gold core of the Au@Ag NPs, respectively.28 Upon addition of H2PtCl6, the silver peak disappears, and the gold peak is red-shifted to 544 nm, indicating at least part of the silver shell was consumed and structural changes in the NPs may take place due to the galvanic replacement reaction between the Au@Ag NPs and H2PtCl6. After the introduction of DMcT, the gold peak is further red-shifted to about 551 nm and obvious aggregation hardly appears in the later centrifugation, which suggests that the sizes of the products further increase in the form of a new shell after DMcT exposure.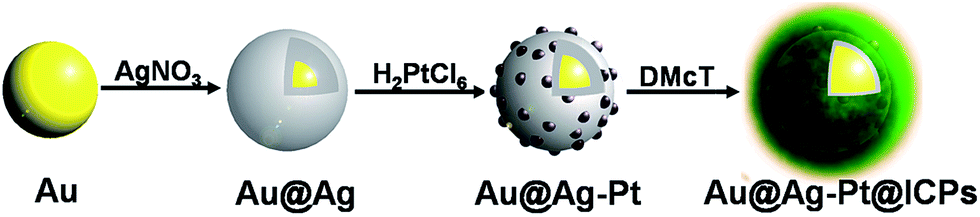 | ||
| Fig. 1 Schematic illustration of the procedure used to prepare Au@Ag–Pt@ICPs NPs. | ||
Fig. 2A and B show a representative TEM image of the Au@Ag–Pt@ICPs NPs and an HRTEM image of the suspected Pt NP area, respectively. The image clearly shows the Au@Ag–Pt@ICP NPs have a threefold-layer core–shell structure that corresponds to a dark Au@Ag (or Au) core, a Pt nanocluster ring, and an obvious layer of an ICP coating of ca. 9 nm in thickness surrounding the Au@Ag–Pt nanostructure. Among them, the Pt nanocluster ring is made up of many small and highly dispersed Pt NPs. In this synthesis, it should be noted that there are almost no bare Au@Ag or Au@Ag–Pt NPs existing, which proved the excellent inclusion ability of the ICPs. Moreover, the EDX analysis (Fig. S2, ESI†) of this sample shows the peaks corresponding to the Au, Ag, Pt, and S elements, which indicates that the sulphur element here should originate from the existence of DMcT-based ICPs. Fig. 2B shows the HRTEM lattice image of the suspected Pt NP area. The lattice spacing of 0.23 nm, marked in Fig. 2B, is consistent with that of Pt (111) planes, and the lattice spacing of 0.20 nm could be attributed to the Pt (100) planes.29 Additionally, the scanning electron microscopy (SEM) image in Fig. 2C further demonstrates the obtained ca. 68 nm Au@Ag–Pt@ICPs NPs are uniform and nearly mono-dispersed. On the other hand, two control experiments on the synthesis of Au@Ag–Pt@ICPs NPs were conducted, and the TEM images of products are shown in Fig. S3.† It was found that the ICP shell could also be formed around “bare” gold NPs (or Au@Ag NPs). In other words, Au@ICPs (or Au@Ag@ICPs) can be easily prepared by simply adding DMcT to the mixture of Au NPs (or Au@Ag NPs) and H2PtCl6. Besides, their dispersibility is also very high. This is superior to methods for fabricating other ICP-based core–shell nanostructures, such as Au@ICPs30 and QDs@ICPs.31 They usually require surface modification with a specific functional group on the Au NPs or QDs; the dispersibility of the core–shell ICP hetero-structures as prepared and the homogeneity of the ICP shells are also not sufficient. Therefore, the approach used in our paper is good and can be easily extended to the synthesis of other ICP-based nanomaterials. It is generally known that NPs can endow polymeric nanocomposites with improved mechanical strength.32 Large numbers of uniformly dispersed Au@Ag NPs and Pt NPs could effectively promote the mechanical strength of the ICPs here, and the stable micro-surrounding for the entrapped enzyme molecules should be favorable to minimize their conformational changes and preserve their bioactivity during utilization and storage of the relevant biosensors. Moreover, the small size and homogeneous loading of the Pt NPs is an important parameter for promoting the performance of our glucose sensors.
 | ||
| Fig. 2 Morphologies of the Au@Ag–Pt@ICPs NPs obtained by successively mixing the prepared Au@Ag NPs (synthesized with 200 μL AgNO3 solution) with 60 μL H2PtCl6 for 30 min, and then 200 μL DMcT solutions for 3 h at room temperature. (A) TEM image; (B) HRTEM image of the suspected Pt NP area inside the Au@Ag–Pt@ICPs NPs; (C) SEM image. XPS spectra of the as-synthesized Au@Ag–Pt@ICPs NPs: (D) survey spectrum and (E) high resolution of Pt spectrum. | ||
DMcT is well-known for its ability to form stable complexes with heavy- and transition-metal ions, like AuCl4−, PtCl62−, PdCl42− and so on.33 Moreover, we have demonstrated that DMcT was able to efficiently react with silver nanostructures at a zero oxidation state and form Ag–DMcT ICPs in our previous research.25 In order to ascertain the composition of the ICPs shell in the Au@Ag–Pt@ICPs NPs, XPS analysis of this sample was conducted. Fig. 2D shows the survey XPS data, which indicates that the as-synthesized Au@Ag–Pt@ICPs NPs contain six elements: Pt, Ag, C, S, N and Cl. Among them, the C, S and N are three main elements of DMcT that should only be present in the DMcT related substances, which is consistent with the EDX result (Fig. S2, ESI†) mentioned above. In addition, the XPS spectrum of Pt4f for the Au@Ag–Pt@ICPs NPs is shown in Fig. 2E, and it can be deconvoluted into two groups of subpeaks labeled 1 and 2 with binding energies of 72.7 and 74.7 eV, respectively. These two peaks can be assigned to Pt2+ and Pt4+, respectively.34 However, the peak at 71.3 eV (corresponding to Pt0) does not exist. Based on this data, it can be inferred that the presence of Pt, C, S and N is indicative of the formation of Pt–DMcT ICPs in the shell of Au@Ag–Pt@ICPs NPs. Additionally, in view of the threefold-layer structure of Au@Ag–Pt@ICPs in Fig. 2A—an Au@Ag core, a Pt nanocluster ring, and an ICP shell from the inside out, the XPS results here should accurately reflect the composition of the ICP shell except for the absence of Pt0. The presence of Ag must be ascribed to Ag+ rather than Ag0 because of the presence of DMcT in the ICP shell.25 Given that there are only two possible kinds of Ag+ ions, namely, AgCl and Ag–DMcT ICPs, and the elemental concentration of Ag is more than that of Cl (Table S1, ESI†), it can be safely inferred that the ICP shell of Au@Ag–Pt@ICPs NPs not only contains Pt–DMcT ICPs but also Ag–DMcT ICPs. In order to confirm this hypothesis, molecular vibrational spectroscopic techniques including FT-IR and Raman spectroscopy were utilized to characterize the Au@Ag–Pt@ICPs NPs as prepared. FT-IR spectra of Au@Ag–Pt@ICPs NPs and pure powder DMcT are shown in Fig. S4.† The stretching vibration peak of S–H of DMcT at 2475 cm−1 is initially observed (Fig. S4-A†),35 but it is absent in the IR spectrum of the Au@Ag–Pt@ICPs NPs (Fig. S4-B†). In addition, the characteristic peak of the stretching vibrations of Pt–N is found at 549 cm−1, but cannot be observed for the pure DMcT, which implies nitrogen coordination.1 In addition, another two characteristic peaks of the stretching vibrations of Ag–S (191 cm−1)36 and Pt–S (390 cm−1)1 are also observed in the Raman spectra of Au@Ag–Pt@ICPs (Fig. S5†). In general, the ICP shell of Au@Ag–Pt@ICPs NPs simultaneously contains Pt–DMcT and Ag–DMcT ICPs structures based on the above results.
3.2 Influence of reactant concentration on the structure of Au@Ag–Pt@ICPs NPs
Given that the ICP shell of Au@Ag–Pt@ICPs NP is made up of a combination of Pt–DMcT and Ag–DMcT ICPs originating from Au@Ag NPs, H2PtCl6 and DMcT, the effects of the amount of DMcT, H2PtCl6 and the composition of Au@Ag NPs on the structure of the products were investigated. In Fig. 3A, there is a thin layer of ca. 9 nm of ICPs coated around the Au@Ag–Pt nanostructure when the volume of DMcT is 200 μL. When the volume of DMcT increases to 600 μL, a much thicker shell (ca. 19 nm) is fabricated (Fig. 3B). However, the thickness of the ICP shell no longer increases (Fig. 3C) if the volume continues to increase to 1 mL. It is reasonable that DMcT in this case may be in excess and no more ICPs can be generated. These results suggest that the ICPs shell can be finely tuned by varying the amount of DMcT. On the other hand, the thickness of the Ag shell of Au@Ag NPs was also investigated. From Fig. 3D–F, it can be clearly seen that the ICP shells thicken and become more and more irregular with an increasing volume of AgNO3. Likewise, it has been demonstrated that the amount of H2PtCl6 has similar effects on Au@Ag–Pt@ICPs NPs, and the results are shown in Fig. S6.† Hence, a conclusion can be conducted that the ICP shell of Au@Ag–Pt@ICPs NPs can be precisely tailored by changing the amount of DMcT and H2PtCl6, as well as the thickness of the Ag shell of the Au@Ag NPs. Given that the loading of the immobilized enzymes is closely related to the thickness of the ICP shell, it is possible to flexibly regulate and control the performance (e.g., detection sensitivity) of the Au@Ag–Pt@ICP-based enzyme electrode as stated later. | ||
| Fig. 3 Influence of the volume of 0.5 mg mL−1 DMcT: (A) 200 μL, (B) 600 μL, (C) 1 mL; the composition of the Au@Ag NPs changed with different volumes of AgNO3: (D) 200, (E) 400, (F) 600 μL on the nanostructures of Au@Ag–Pt@ICPs NPs. | ||
3.3 Electrochemical performance for glucose detection
The electrochemical behavior of the modified electrodes was first investigated by cyclic voltammetry (CV) using Fe(CN)63−/4− as the redox marker to ensure the successful binding of GOx to the Au@Ag–Pt@ICPs. CVs were obtained with bare Pt, Au@Ag–Pt@ICPs/Pt and Au@Ag–Pt@ICPs–GOx/Pt electrodes in 0.1 M KCl with 1 mM of Fe(CN)63−/4− (Fig. S7†). A pair of one electron quasi-reversible redox waves can be observed at the bare Pt electrode, and the peak separation between the anodic and cathodic waves was about 84 mV. After the Au@Ag–Pt@ICPs were modified on the bare Pt electrode, the peak separation slightly increased to about 89 mV and the peak current slightly decreased. These phenomena indicated the formation of an Au@Ag–Pt@ICPs film on the electrode surface and the weak hindrance effect on the redox couples due to the insulating ICP shell. Furthermore, when the Au@Ag–Pt@ICPs–GOx was modified, the peak separation increased significantly, and the peak current decreased significantly in comparison to the Au@Ag–Pt@ICPs, which demonstrated the GOx was successfully immobilized on/in the Au@Ag–Pt@ICPs–GOx NPs.The electrochemical activity of the as-prepared Au@Ag–Pt@ICPs–GOx based electrode was further detected by CV experiments. Fig. 4 shows the CV behaviors of the different material modified electrodes (fabricated under optimized conditions (Table S2†)) in a 0.1 M PBS solution with or without the presence of glucose. No oxidation peak was obtained for the Au@Ag–Pt@ICPs–GOx fabricated electrode in the blank PBS without glucose (Fig. 4A-4, black line). Nevertheless, after the addition of 5.0 mM glucose, a prominent anodic peak (+0.48 V) with significant peak current appeared (Fig. 4A-4, red line). Of the several CV curves in Fig. 4A, which correspond to the different ICP-based material fabricated electrodes in 5.0 mM glucose, including (1) ICPs–GOx, (2) Au–ICPs–GOx, (3) Au@Ag@ICPs–GOx and (4) Au@Ag–Pt@ICPs–GOx, Fig. 4A-4 showed the highest current value, suggesting that Au@Ag–Pt@ICPs–GOx has the best eletrocatalytic activity for the detection of glucose. TEM characterizations of these four ICP-based materials were further conducted (Fig. 4B). In the case of the ICPs–GOx (very little product could be collected), the film presented a loose and porous structure ascribed to the existence of pure ICPs (Fig. 4B-1), which was similar to a previous report.1 In the case of Au–ICPs–GOx, only bare Au NPs were obtained and Pt–DMcT ICPs, which can be used to load enzymes that no longer coat around the Au NP core (Fig. 4B-2) in comparison to the case without GOx (Fig. S3-A†). In the case of the Au@Ag@ICPs–GOx, typical ICP-based core–shell nanostructures were produced (Fig. 4B-3). In this case, the high structural tailorability of ICP shells could increase the amount of immobilized GOx, and the inside Au@Ag NPs could act as good electric conductors to improve the electrochemical sensing ability.37 It is worth noting that new nanostructures with urchin-like morphologies were fabricated in the case of Au@Ag–Pt@ICPs–GOx (Fig. 4B-4). Compared to the case of Au@Ag@ICPs–GOx, this more loose and porous structure not only favored the higher density immobilization of the GOx and the faster penetration of water-soluble molecules (e.g. glucose and H2O2) but it also could catalyze the decomposition of H2O2 more quickly with the aid of the small Pt NPs (Fig. 2A). All of this is responsible for the high performance of our Au@Ag–Pt@ICPs–GOx biosensor.
 | ||
| Fig. 4 (A) CVs of the electrodes: (1) ICPs–GOx/Pt, (2) Au–ICPs–GOx/Pt, (3) Au@Ag@ICPs–GOx/Pt and (4) Au@Ag–Pt@ICPs–GOx/Pt with (red line) and without (black line) 5.0 mM glucose in 0.1 M PBS (pH 7.0); scan rate = 50 mV s−1. The enzyme electrodes were fabricated under optimized conditions. (B) TEM images of (1) ICPs–GOx, (2) Au–ICPs–GOx, (3) Au@Ag@ICPs–GOx and (4) Au@Ag–Pt@ICPs–GOx. | ||
The amperometric glucose-biosensing responses and the calibration curves of the four enzyme electrodes as mentioned above are shown in Fig. 5, and the values of sensitivity (S), linear detection range (LDR), and limit of detection (LOD, S/N = 3) are listed in Table S3.† It is clearly seen that rapid responses to glucose could be achieved for all of the electrodes except for the Au@Ag@ICPs–GOx/Pt electrode (Fig. 5A). The average response time for glucose on our Au@Ag–Pt@ICPs–GOx/Pt electrode (reaching 95% of its steady-state value) was as short as 4 s, which is smaller than previously reported values for many polymeric nanocomposite-based biosensors.11,38,39 The fast response of our sensor may be attributed to the loose and porous 3D urchin-like nanostructures (Fig. 4B-4) that facilitate the diffusion of the glucose and the H2O2. Moreover, the sensitivity calculated from the linear portion of the calibration of Au@Ag–Pt@ICPs–GOx/Pt is as high as 82.1 μA mM−1 cm−2, which is 5.2, 16.1, and 3.1 fold that of the ICPs–GOx/Pt, Au–ICPs–GOx/Pt and Au@Ag@ICPs–GOx/Pt (Table S3†), respectively. Furthermore, the LOD of Au@Ag–Pt@ICPs–GOx/Pt is almost two orders of magnitude lower than that of ICPs–GOx/Pt (Table S3†).
 | ||
| Fig. 5 Chronoamperometric responses to successive additions of glucose (A) and the calibration curves (B) on (1) ICPs–GOx/Pt, (2) Au–ICPs–GOx/Pt, (3) Au@Ag@ICPs–GOx/Pt and (4) Au@Ag–Pt@ICPs–GOx/Pt at 0.7 V versus SCE in PBS (pH 7.0). The enzyme electrodes were fabricated under optimized conditions. The linearly regressed lines are also shown. | ||
The sensitivity of our Au@Ag–Pt@ICPs–GOx/Pt biosensor is also notably higher than certain ICP-based glucose electrochemical sensors and Pt-based glucose biosensors (Table 1). On the other hand, we also pre-synthesized Pt NPs, which were first mixed with Au NPs or Au@Ag NPs before adding the mixture of H2PtCl6 and DMcT to cast-coat the enzyme films on Pt electrodes by a similar method (Au-pre-synthesized Pt NPs–ICPs–GOx/Pt and Au@Ag-pre-synthesized Pt NPs–ICPs–GOx/Pt, respectively). In these processes, we found that the metal NPs could not be easily included into the ICPs, similar to the phenomena of Au–ICPs–GOx (Fig. 4B-2), and a very small amount of products with very little loaded GOx could be collected. Electrochemical experiments (Fig. S8†) further proved that these two electrodes showed a much poorer performance than the Au@Ag–Pt@ICPs–GOx/Pt electrode. All of the above discussion suggested that the high sensitivity of our biosensor may result from a synergy effect of the noble metal NPs and the 3D porous matrix of the ICPs in the urchin-like nanostructures of the Au@Ag–Pt@ICPs–GOx, which not only allowed for the high load/activity of the immobilized biorecognition molecules but also allowed for excellent catalytic activity for the oxidation of the H2O2 generated during the enzymatic reaction.
| Electrodes | S (μA cm−2 mM−1) | Response time (s) | LDR (mM) | LOD (μM) | Km (mM) | Ref. |
|---|---|---|---|---|---|---|
| Pt/copper frameworks | 9.62 | — | 1–11 | 0.385 | — | 40 |
| GOx/Pt-DENs/PANI/CNT/Pt | 42 | 5 | 0.001–12 | 0.5 | — | 41 |
| GOx-PoAP/polypyrrole–Pt NPs/GCE | 9.9 | ∼7 | 0.0015–13 | 0.45 | 24 | 39 |
| PtAuPd/carbon nanotubes | 11.24 | — | 0–10 | 2.9 | — | 42 |
| GOx–CS–SiO2/Pt/MWNTs/GCE | 59 | 5 | 0.001–23 | 1 | 14 | 43 |
| GOx/Pt NP/PANI/Pt | 96.1 | 3 | 0.01–8 | 0.7 | 0.57 | 29 |
| Pt/graphene | 1.21 | 3 | 1–25 | 30 | — | 44 |
| Pt/carbon microspheres | 15.4 | — | — | — | — | 45 |
| Pt–Ni alloy nanotube | 124.17 | — | Up to 13.5 | 32 | — | 46 |
| MEBCs2/Au | 52 | — | 0.075 × 10−3 to 0.7 | 0.075 | 1 | |
| ICPs–GOx/Pt | 15.8 | — | 0.005–2.225 | 2.7 | 3.51 | This work |
| Au@Ag–Pt@ICPs–GOx/Pt | 82.1 | 4 | 0.0005–3.325 | 0.06 | 0.28 | This work |
The apparent Michaelis–Menten constant (Km) can be calculated from the electrochemical version of the Lineweaver–Burk equation:
| 1/Iss = 1/Imax + Km/Imax × C | (1) |
Different aspects regarding the characteristics of our Au@Ag–Pt@ICPs–GOx biosensor were further evaluated. The repeatability and reproducibility were conducted at a glucose concentration of 1 mM. The relative standard deviation (RSD) for six parallel measurements is 3.8% (repeatability), and the RSD for six freshly prepared electrodes is 4.9% (reproducibility). The storage stability was studied by making measurements with one-week intervals. We found that the amperometric response of the biosensor to 1.0 mM glucose was almost constant after 9 weeks and decreased to approximately 90% after 14 weeks (Fig. 6A), which is much better than certain ICP-based electrochemical sensors and noble metal NP-based glucose sensors (Table S4†). The excellent long-term stability of our proposed biosensors can probably be attributed to the one-pot fabrication of the bio-electrodes and the synergism effect of metal NPs and ICPs. The electrodes were fabricated under mild conditions with minimal damage to the enzyme molecules. Additionally, the excellent biocompatibility and mechanical stabilities of Au, Ag, Pt, as well as the high adsorption/encapsulation capability and unique porous structure of ICP materials contributed to maintaining the bioactivity of the enzyme molecules immobilized on the electrodes.
 | ||
| Fig. 6 (A) The fabrication storability of the Au@Ag–Pt@ICPs–GOx/Pt electrode (at least three parallel determinations for each). (B) Contrastive study of the effect of temperature on the activities of Au@Ag–Pt@ICPs–GOx/Pt (black line) and ICPs–GOx/Pt (red line) electrodes. | ||
To further study the effect of metal NPs on the enzymatic activity of GOx, the influence of temperature on the relative activities of Au@Ag–Pt@ICPs–GOx/Pt and ICPs–GOx/Pt electrodes was also investigated between 20 and 70 °C (Fig. 6B). It can be clearly found that the introduction of metal NPs greatly improved our proposed biosensor's thermo-stability. The enzymatic activity of the Au@Ag–Pt@ICPs–GOx/Pt electrode increased when the temperature increased from 20 to 50 °C and reached a maximum response at approximately 50 °C. However, the electrode can still maintain over 90 percent efficiency even at 60 °C. However, the enzymatic activity of the ICPs–GOx/Pt electrode decreases dramatically from 40 °C due to denaturation of the enzyme. It should be noted that the temperature (60 °C) to retain enough GOx enzymatic activity is also pretty high in comparison with previous reported works.9,39,49 The excellent thermo-stability of our biosensor could be attributed to the metal NPs in the urchin-like structure of Au@Ag–Pt@ICPs–GOx, which greatly improved the mechanical strength of the ICPs as mentioned above.
On the other hand, the influence of some potential interfering substances on the current response of glucose was tested on our Au@Ag–Pt@ICPs–GOx/Pt biosensor. Fig. S9† represents the chronoamperometric responses of glucose (3 mM) in the presence of AA (0.1 mM), fructose (0.1 mM) and L-cysteine (0.05 mM). It can be seen that the above interferences did not influence the current response of glucose from the proposed biosensor. Hence, the Au@Ag–Pt@ICPs–GOx/Pt electrode exhibited a good selectivity of glucose over these molecules with similar structures. To illustrate the feasibility of our proposed biosensor in a biologically relevant matrix, the determination of glucose in human serum samples is also conducted. Three fresh blood samples obtained from hospitalized patients were first diluted 100-fold by a PBS buffer to yield testing sample solutions of pH 7.0 before determination. In addition, a recovery test was conducted. All the data are summarized in Table 2. The biosensor's results agreed well with the reference ones that were obtained by the GOx-peroxidase method in a local hospital, and a good recovery was also obtained. Thus, it can be concluded that the developed sensor performs very well in the detection of glucose in serum samples.
| Blood serum sample | Determined in hospital (mM) | Determined by biosensor (mM) | RSD (%) | Standard added (mM) | Found (mM) | Recovery (%) |
|---|---|---|---|---|---|---|
| 1 | 5.24 | 5.01 | −4.39 | 5.00 | 4.95 | 99.0 |
| 2 | 7.62 | 7.41 | −2.75 | 5.00 | 4.67 | 93.4 |
| 3 | 6.20 | 6.53 | +5.32 | 5.00 | 5.13 | 102.5 |
4. Conclusion
In summary, for the first time, a novel and controllable triplex noble metal nanoparticle (NP)-mixed infinite coordination polymer (ICP) core–shell NPs (Au@Ag–Pt@ICPs) was successfully fabricated based on the galvanic replacement reaction of Au@Ag NP and H2PtCl6, as well as the coordination reactions of 2,5-dimercapto-1,3,4-thiadiazole and two kinds of metal ions source, H2PtCl6 and nanoscale metallic Ag. The strategy presented here is simple, one-pot and can be fulfilled in aqueous solution at room temperature. Importantly, no surface functionalization for NPs is needed, and the products' dispersibility and the ICP shells' homogeneity are also very good. Moreover, due to the excellent conductivity, mechanical stabilities, catalytic activity and biocompatibility of the metal NPs as well as the high structural tailorability of ICP materials, the Au@Ag–Pt@ICPs NPs as prepared can in situ immobilize enzymes (e.g., glucose oxidase) with a high load/activity and exhibit obvious electrocatalysis/nano-enhancement effects for biosensing of glucose. Specifically, the as prepared glucose biosensors exhibit excellent performance with a much higher sensitivity, a much better long-term stability and thermal stability, and a very lower Michaelis–Menten constant compared to ICP-based biosensors prepared only with ICP materials and reported analogues. We believe the new strategy proposed here may be applied to prepare many other multi-functionalized ICP-based nanomaterials for wide applications in the fields of biosensing, biofuels, biocatalysis, environmental monitoring, human health, and so on.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 81471696, 21475100, 41273093 and 21175101), Natural Science Foundation of Hubei Province of China (No. 2014CFA002), and Foundation of China Geological Survey (No. 12120113015200).Notes and references
- Y. C. Fu, P. H. Li, L. J. Bu, T. Wang, Q. J. Xie, J. H. Chen and S. Z. Yao, Anal. Chem., 2011, 83, 6511 CrossRef CAS PubMed
.
- L. Li, F. F. Gao, J. Ye, Z. Z. Chen, Q. L. Li, W. Gao, L. F. Ji, R. R. Zhang and B. Tang, Anal. Chem., 2013, 85, 9721 CrossRef CAS PubMed
- R. Weiss, Y. Yegorchikov, A. Shusterman and I. Raz, Diabetes Technol. Ther., 2007, 9, 68 CrossRef CAS PubMed
- M. J. Tierney, J. A. Tamada, R. O. Potts, L. Jovanovic, S. Garg and C. R. Team, Biosens. Bioelectron., 2001, 16, 621 CrossRef CAS PubMed
- J. Wang, Chem. Rev., 2008, 108, 814 CrossRef CAS PubMed
- M. Hartmann, Chem. Mater., 2005, 17, 4577 CrossRef CAS
- S. J. Guo, S. J. Dong and E. K. Wang, Small, 2009, 5, 1869 CrossRef CAS PubMed
- C. Chen, Y. C. Fu, C. H. Xiang, Q. J. Xie, Q. F. Zhang, Y. H. Su, L. H. Wang and S. Z. Yao, Biosens. Bioelectron., 2009, 24, 2726 CrossRef CAS PubMed
- C. Y. Hu, D. P. Yang, F. J. Zhu, F. J. Jiang, S. Y. Shen and J. L. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 4170 CAS
- Y. C. Fu, C. Zou, Q. J. Xie, X. H. Xu, C. Chen, W. F. Deng and S. Z. Yao, J. Phys. Chem. B, 2009, 113, 1332 CrossRef CAS PubMed
- Y. C. Fu, P. H. Li, Q. J. Xie, X. H. Xu, L. H. Lei, C. Chen, C. Zou, W. F. Deng and S. Z. Yao, Adv. Funct. Mater., 2009, 19, 1784 CrossRef CAS
- S. Cosnier, C. Gondran, J. C. Watelet, W. D. Giovani, R. P. M. Furriel and F. A. Leone, Anal. Chem., 1998, 70, 3952 CrossRef CAS
- F. F. Han, X. Qi, L. Y. Li, L. J. Bu, Y. C. Fu, Q. J. Xie, M. L. Guo, Y. B. Li, Y. B. Ying and S. Z. Yao, Adv. Funct. Mater., 2014, 24, 5011–5018 CrossRef CAS
- L. Wang, Q. Y. Zhang, S. L. Chen, F. G. Xu, S. H. Chen, J. B. Jia, H. L. Tan, H. Q. Hou and Y. H. Song, Anal. Chem., 2014, 86, 1414 CrossRef CAS PubMed
- Y. G. Liu, X. M. Feng, J. M. Shen, J. J. Zhu and W. H. Hou, J. Phys. Chem. B, 2008, 112, 9237 CrossRef CAS PubMed
- M. R. Romero, F. Garay and A. M. Baruzzi, Sens. Actuators, B, 2008, 131, 590 CrossRef CAS
- P. C. Huang, J. J. Mao, L. F. Yang, P. Yu and L. Q. Mao, Chem.–Eur. J., 2011, 17, 11390 CrossRef CAS PubMed
- X. L. Lu, H. J. Cheng, P. C. Huang, L. F. Yang, P. Yu and L. Q. Mao, Anal. Chem., 2013, 85, 4007 CrossRef CAS PubMed
- Y. J. Guo, Y. J. Han, S. M. Shuang and C. Dong, J. Mater. Chem., 2012, 22, 13166 RSC
- U. Lad, G. M. Kale and R. Bryaskova, Anal. Chem., 2013, 85, 6349 CrossRef CAS PubMed
- H. H. Zhou, H. Chen, S. L. Luo, J. H. Chen, W. Z. Wei and Y. F. Kuang, Biosens. Bioelectron., 2005, 20, 1305 CrossRef CAS PubMed
- A. Heller, Acc. Chem. Res., 1990, 23, 128 CrossRef CAS
- S. Lee, B. S. Ringstrand, D. A. Stone and M. A. Firestone, ACS Appl. Mater. Interfaces, 2012, 4, 2311 CAS
- X. E. Jia, G. Z. Hu, F. Nitze, H. R. Barzegar, T. Sharifi, C. W. Tai and T. Wågberg, ACS Appl. Mater. Interfaces, 2013, 5, 12017 CAS
- L. H. Wang, A. G. Shen, X. C. Li, Y. Zeng, X. D. Zhou, R. M. Richards and J. M. Hu, RSC Adv., 2014, 4, 34294 RSC
- B. Y. Wu, S. H. Hou, F. Yin, J. Li, Z. X. Zhao, J. D. Huang and Q. Chen, Biosens. Bioelectron., 2007, 22, 838 CrossRef CAS PubMed
- R. Polsky, R. Gill, L. Kaganovsky and I. Willner, Anal. Chem., 2006, 78, 2268 CrossRef CAS PubMed
- A. G. Shen, L. F. Chen, W. Xie, J. C. Hu, A. Zeng, R. Richards and J. M. Hu, Adv. Funct. Mater., 2010, 20, 969 CrossRef CAS
- D. Y. Zhai, B. R. Liu, Y. Shi, L. J. Pan, Y. Q. Wang, W. B. Li, R. Zhang and G. H. Yu, ACS Nano, 2013, 7, 3540 CrossRef CAS PubMed
- R. Nishiyabu, N. Hashimoto, T. Cho, K. Watanabe, T. Yasunaga, A. Endo, K. Kaneko, T. Niidome, M. Murata, C. Adachi, Y. Katayama, M. Hashizume and N. Kimizuka, J. Am. Chem. Soc., 2009, 131, 2151 CrossRef CAS PubMed
- R. Nishiyabu, C. Aimé, R. Gondo, K. Kaneko and N. Kimizuka, Chem. Commun., 2010, 46, 4333 RSC
- R. Gangopadhyay and A. De, Chem. Mater., 2000, 12, 608 CrossRef CAS
- M. R. Gajendragad and U. Aqarwa, Z. Anorg. Allg. Chem., 1975, 415, 84 CrossRef CAS
- W. Eberhardt, P. Fayet, D. Cox, Z. Fu, A. Kaldor, R. Sherwood and D. Sondericker, Phys. Rev. Lett., 1990, 64, 780 CrossRef CAS PubMed
- P. Ortega, L. R. Vera and M. E. Guzmán, Macromol. Chem. Phys., 1997, 198, 2949 CrossRef CAS
- V. T. Joy and T. K. K. Srinivasan, J. Raman Spectrosc., 2001, 32, 785 CrossRef CAS
- C. Y. Hu, D. P. Yang, K. Xu, H. M. Cao, B. N. Wu, D. X. Cui and N. Q. Jia, Anal. Chem., 2012, 84, 10324 CrossRef CAS PubMed
- D. Wan, S. J. Yuan, G. L. Li, K. G. Neoh and E. T. Kang, ACS Appl. Mater. Interfaces, 2010, 2, 3083 CAS
- J. Li and X. Q. Lin, Biosens. Bioelectron., 2007, 22, 2898 CrossRef CAS PubMed
- Y. L. Hu, X. H. Niu, H. L. Zhao, J. Tang and M. B. Lan, Electrochim. Acta, 2015, 165, 383 CrossRef CAS
- L. H. Xu, Y. H. Zhu, X. L. Yang and C. Z. Li, Mater. Sci. Eng., C, 2009, 29, 1306 CrossRef CAS
- B. Singh, E. Dempsey and F. Laffir, Sens. Actuators, B, 2014, 205, 401 CrossRef CAS
- Y. J. Zou, C. L. Xiang, L. X. Sun and F. Xu, Biosens. Bioelectron., 2008, 23, 1010 CrossRef CAS PubMed
- G. Chang, H. H. Shu, Q. W. Huang, M. Oyama, K. Ji, X. Liu and Y. B. He, Electrochim. Acta, 2015, 157, 149 CrossRef CAS
- X. H. Niu, H. L. Zhao, M. Lan and L. Zhou, Electrochim. Acta, 2015, 151, 326 CrossRef CAS
- Y. Sun, H. Y. Yang, X. H. Yu, H. W. Meng and X. H. Xu, RSC Adv., 2015, 5, 70387 RSC
- T. Tsuchida and K. Yoda, Enzyme Microb. Technol., 1981, 3, 326 CrossRef CAS
- B. E. P. Swoboda and V. J. Massey, J. Biol. Chem., 1965, 240, 2209 CAS
- Z. Y. Wang, S. N. Liu, P. Wu and C. X. Cai, Anal. Chem., 2009, 81, 1638 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Additional characterization data of Au@Ag–Pt@ICPs NPs such as UV-vis, EDX, IR spectra, Raman spectra analysis and TEM images; more data on the optimization and evaluation of the Au@Ag–Pt@ICPs based biosensors. See DOI: 10.1039/c6ra15293g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |