
Structural and conformational insights into SOX2/OCT4-bound enhancer DNA: a computational perspective†
Muhammad Ayaz Anwar,
Dhanusha Yesudhas,
Masaud Shah and
Sangdun Choi*
Department of Molecular Science and Technology, Ajou University, Suwon 443-749, Korea. E-mail: sangdunchoi@ajou.ac.kr; Fax: +82-31-219-1615; Tel: +82-31-219-2600
First published on 15th September 2016
Abstract
The potential role of sex determining region Y-box 2 (SOX2) and octamer-binding transcription factor 4 (OCT4) are increasingly discussed in stem cell maintenance either in the context of iPSCs (induced pluripotent stem cells) generation or cancer stem cell growth. These proteins bind to the enhancer and drive the transcription of a multitude of other factors that facilitate stem cell propagation. Here, we elucidated the mechanism of changes in DNA shape and the precise role of the interaction with the proteins, which is necessary to manipulate this ternary complex. Besides bending the DNA, SOX2 drove the DNA into the A-form, whereas OCT4 preferentially shaped DNA into a B-like conformation. SOX2 binding expanded the minor groove with simultaneous shrinkage of the major grove. Greater fluctuation in the DNA and bound proteins was observed after disruption of the protein–protein interaction. Dynamic cross-correlation of DNA atoms was found to be variable, and entropy of DNA atoms from DNA-wild-type-SOX2/OCT4 (DNAWT) was the lowest among the various complexes. Moreover, essential dynamics-based conformational analysis revealed vivid conformational variation both in DNA alone and in protein bound complexes. Physical parameters such as the diffusion coefficient and dipole moment were also substantially different for DNA from the DNAWT complex. Taken together, our results establish a link between protein–protein and protein–DNA interactions, which will facilitate devising various strategies to modulate this complex in order to regulate the transcription of various proteins.
Introduction
Transcription factors can specifically bind to a short stretch of DNA located in the regulatory region of a gene. This protein–DNA interaction depends on various factors such as DNA sequence, DNA structural configuration and domains present in proteins. Moreover, evolution has placed a large number of transcription factor-binding sites in close proximity: a phenomenon known as the enhanceosome, which enhances specificity and cooperative binding to such a site. This cooperation enhances the DNA-binding specificity, and the partner proteins modulate each other's affinity.1,2 Further regulatory control has been achieved by evoking a certain structural alteration in DNA after binding of the first protein, which allosterically affects the binding of another protein even without protein–protein interaction.3–6 Despite recent insights into enhanceosome assembly,7,8 the structural and mechanistic details of how signal is perpetuated between the binding sites and how protein binding improves specificity of another protein's binding have long been poorly studied topics.Aside from sequence-specific binding, DNA has minor and major grooves that may also be recognized by proteins in a sequence-independent manner.9 For instance, the OCT4 (octamer-binding transcription factor 4) protein, which belongs to the POU family, is composed of two distinct domains, a POU-specific domain (POUS) and the POU homeodomain (POUHD), which are connected by a flexible linker. The helix of POUS and the N-terminal part of POUHD can bind to major groove and minor groove of DNA, respectively, thereby establishing protein–DNA contacts for subsequent transcription.10 The characteristic 8-mer pattern for OCT4 recognition is ATGC(A/T)AAT, where POUS and POUHD bind to first and second half of this consensus sequence consecutively.11 This transcription factor is vital for induced stem cell pluripotency and performs its function in combination with other proteins, mostly SOX2, as determined by whole-genome chromatin immunoprecipitation analysis.12–14 The binding sites for OCT4 and SOX2 are juxtaposed in the regulatory region of many proteins, which are necessary to generate, maintain and propagate stem cells.
The consensus sequence of the binding site for the SOX (Sry-related HMG box) family is CTTTGT,15 with minor variations among cell types. The relative positions of SOX2 and OCT4 binding sites on DNA may have 0- or 3-bp separation,10 whereas OCT4 can bind to SOX17 in a shortened motif (lacking 1 bp); however, this combination lacks stem cell induction. A mutated variant of SOX17 can bind to OCT4 on canonical motif and can induce cell transformation.16
Both OCT4 and SOX2 bind to DNA in a cooperative manner, and POUS of OCT4 forms an interface with SOX2 upon binding; however, SOX2 facilitates and influences the OCT4 binding in vivo,13,17,18 and in most cases, SOX2 is the first to bind to DNA.13 In contrast, it has also been reported that OCT4 alone can bind to DNA during the initial stage of reprogramming, thus pointing to another possible mechanism of DNA recognition in a cell context-dependent manner.19 Although OCT4 and SOX2 can bind to these sites separately, in many instances, only their association activates transcription efficiently.20,21 The direct interaction between OCT4 and SOX2 is DNA dependent18 and involves the POUS helix α1 and the HMG helix α3. These observations revealed different orientation and spacing between these proteins and therefore uncovered the plausible mechanism of interaction of OCT4 and SOX2 proteins that is required for effective iPSCs (induced pluripotent stem cells) induction.22,23 To further test whether members of the SOX and OCT families evolved features to cooperatively target specific enhancer elements, a global assessment of the SOX/OCT pairing profile is highly desirable. Besides, the role of SOX2 and OCT4 are increasingly acknowledged in cancerous cells and cancer stem cells referring their vital role in cell propagation. The frequent overexpression of these proteins in various cancers and their target genes are evident in literature.24,25
Resolution of atomic structures of OCT4 and SOX2 proteins has shed light on their binding preferences;10,26,27 however, there are no studies on how the binding of one protein to DNA can facilitate the binding of the other protein, or what conformational alterations in DNA perpetuate such allosteric mechanisms. There are many studies on protein–DNA interaction, particularly, studies that are focused on protein flexibility and adaptational behavior. In contrast, how DNA modifies its structure and undergoes a conformational change (that provides a feasible framework for the whole complex to perform its functions) has long been a neglected topic. Conformational changes in DNA play a critical role in protein–DNA interactions that result in replication, transcription and DNA modification and thus regulate expression of various genes in a tissue- and condition-dependent manner. Accordingly, this study was designed to explore further details of conformational alteration in DNA allostery by means of molecular dynamics (MD) simulations.
Results
1. Dynamics of protein–DNA complexes
A better understanding of the dynamics of SOX2/OCT4 over DNA is helpful to design additional methods to modify and regulate this complex in a precise manner; these considerations prompted this study (ESI Fig. S1†). All our systems here vary in individual components possibly to encompass the DNA variability and dynamics during MD simulations. These systems have been created through various tools and procedures including homology modelling, rotamer based amino acid substitution, a DNA structure prediction and modification. All DNA molecules and protein–DNA complexes were stable throughout the simulation (250 and 100 ns of repeated simulation). The simulations have been performed using leap-frog algorithm that solves the Newton's equation of motion. The terminals of DNA were free in all cases for analysis of the DNA parameters and bending propensity because SOX2 is known to bend DNA. Seven systems were generated, DNA was extracted from the crystal structure 1GT0 (DNACRY); with the same sequence, a perfect B-DNA was created using 3DAART (DNASYN), crystal structure of DNA and SOX2 (DNASOX) and of DNA and OCT4 (DNAOCT), the whole crystal structure with WT SOX2 (DNAWT), mutated SOX2 (R113E; DNAMUT), and finally a non-specific complex that was generated by mutating the DNA to a non-specific sequence (DNANS). DNASYN and DNACRY represent DNA alone (without any protein). The number of water molecules that were closer than 4 Å to DNA was slightly higher for free DNAs (32.45 per base pair of DNA), intermediate for DNASOX and DNAOCT individual complexes (28.90), lower for DNAMUT and DNANS (26.3), and the lowest for DNAWT (25.66). These water molecules can easily be interchanged with those from bulk water.The relative movement of the protein–DNA complex was estimated by root mean square deviation (RMSD) that measures the structural similarity between two structures after overlapping them, and we found that tetrameric complexes (DNAWT, DNAMUT and DNANS) are highly stable; however, complexes DNASYN, DNACRY, DNASOX and DNAOCT fluctuated slightly (Fig. 1A). DNASYN and DNACRY fluctuated slightly more than other systems that are having SOX2/OCT4 proteins; this effect may influence the overall RMSD value in complexes (ESI Fig. S2A and S2B†). The plausible reason includes lack of any protein bound to these systems. Moreover, these systems comprised of only DNAs, which are held-together by non-covalent H-bonds that has comparatively less influence on the structure to reduce the fluctuations. The number of hydrogen bonds (H-bonds) between two DNA strands was quite stable over time (ranged from 55 to 57), except for DNANS, which showed on average 52.3 H-bonds per frame (Fig. 1B). This result corresponds to the potential number of H-bonds that reinforce the force field capability to reproduce the empirical values in that particular sequence. The number of H-bonds between protein and DNA was proportional to the size of the protein, i.e., SOX2 is a smaller protein with a smaller number of H-bonds (ESI Fig. S3†). Nonetheless, the number of H-bonds between DNA and wild-type (WT) proteins (SOX2/OCT4) was slightly higher than in DNAMUT and DNANS, indicating stability of DNAWT. Moreover, higher energy was required to reform the H-bonds within dsDNA in DNASYN, followed by DNACRY, DNASOX, DNAWT and DNANS as per Luzar and Chandler's description of H-bond kinetics.28 DNAOCT and DNAMUT required the least energy for reforming of DNA H-bonds (ESI Table S1†). DNASYN represents the perfect B-form of DNA and should require more energy for breaking its H-bonding network. Finally, a list of residues that participate in H-bond interaction has been provided to highlight the role of various amino acids and base-pairs in protein–DNA interaction (ESI Table S2†).
 | ||
| Fig. 1 Dynamic characteristics of protein–DNA complexes. (A) Root mean square deviation (RMSD) was measured for the backbone and heavy atoms only in the protein and DNA, respectively. Free DNAs or DNA bound to a single protein showed greater fluctuations but within limits. (B) The number of hydrogen bonds (H-bonds) within DNA strands, considering the criteria for H-bonds as the donor–acceptor distance of 3.5 Å and the hydrogen-donor–acceptor bond angle of 30° or less, and the values are given as the running average of 100 over the trajectory for better understanding. Root mean square fluctuations (RMSF) for (C) DNA and for (D) protein per residue. | ||
Root mean square fluctuation (RMSF), which provides a measure of the atomic mobility, was employed to study conformational changes in the protein and DNA at the atomic level independently (Fig. 1C and D). RMSF for DNA was evaluated per base; this assay revealed that DNAWT displayed the least fluctuation, whereas DNAMUT, DNANS and DNAOCT showed intermediate fluctuations. Other systems, DNASYN, DNACRY and DNASOX, showed greater fluctuation. It is relevant to mention that DNAWT and DNAMUT are identical except for one amino acid, yet the mutated complex showed greater fluctuation. Similarly, proteins from the DNAWT system showed intermediate fluctuation; however, proteins alone from DNASOX and DNAOCT showed less fluctuation than did proteins from DNAWT. Proteins from DNAMUT showed greatest fluctuations particularly in SOX2 and in the POUHD region of OCT4; this result is suggestive of a crucial interaction between SOX2 and OCT4. Notably, in DNANS, SOX2 and POUHD showed greater fluctuation, whereas POUS fluctuation was similar to that of DNAWT. This relative fluctuation of different complexes is critical for stable protein–DNA binding.
In protein–DNA interactions, the role of various positively charged amino acids is crucial and has been evaluated. In DNASOX complex, the terminal loops of SOX2 (N-terminal D39-N46 and C-terminal H105-K117) showed an abrupt binding and dissociation with DNA apart from other amino acids that established a continuous interaction. Moreover, various residues from N-terminal loop (K42, R43, M45), α1 (first α-helix of the protein) (F48, M49, R53/56/57), α2 (S72, K73, G76, W79), α3 (R98) and multiple positively charges residues from C-terminal loop kept a continuous interaction with surrounding DNA residues. In DNAOCT, various amino acids were interacting with DNA through noncovalent bonding. However, the prominent amino acids include R157, T163, Q164, S180, Q181, T182, R186, K195 and N196 from POUS domain, and R242, K254, R275, N280 and Q283 from POUHD domain that were consistent in their interactions. The intermediate loop connecting these domains has shown to heavily interact with DNA since it was wrapping around the DNA. Moreover, this intermediate loop has shown interactions with other amino acids of POUS and POUHD domains, prominently, multiple residues of α2 (G172, V173) and α5 (A223, E224, L226, V227) of the POUS domain, and the α1 (R240, V241, R242 and less frequently L245) of POUHD domain. It has been observed that W277 from POUHD was also in a consistent interaction with this loop due to its large hydrophobic side chain.
In DNAWT, the C-terminal loop of SOX2 (H105-K117) was making extensive bonding network not only with DNA but also with POUS of OCT4. The consistently interacting amino acids from OCT4 are form α1 and α2 of POUS domain, such as K154, T163 and D166. Besides, substantial interactions have also been observed with K151, I158 and G161 of POUs domain. This loop has shown interaction with initial arginine residues (R40 and R43) of SOX2 as well. As it has been stated that this loop has shown moderately stable association in DNASOX complex, it was highly stable in DNAWT and no dissociation event has been observed. This is mainly due to the presence of OCT4 that helps to stabilize the complex. The amino acids that interacted with DNA were largely the same as have been reported in individual complexes.
In case of DNAMUT, SOX2 loop (H105-K117) could not establish the interaction with K154, while the interaction with T163 and D166 were preserved. Moreover, most of the interactions as observed in DNAWT were lost. This SOX2 C-terminal loop has unstable interaction with α5 of POUS and the initial region of intermediate loop of OCT4 (E219-T225). It is apparent that by mutating a positive residue with a negative residue (R113E), the stability effect due to electrostatic interaction and arginine intercalation has been wiped out, which has reduced its affinity with DNA. In DNANS complex, SOX2 C-terminal loop significantly displaced from the minor groove of DNA that abolished most of the native contacts (except T163 and D166). Similar to DNAMUT complex, this loop was establishing more contacts with α5 of POUs domain and the intervening loop that connects two POU domains. Moreover, this C-terminal loop showed a few interactions with α3 of SOX2 (R98, M102). In DNANS complex, SOX2 and OCT4 lack specific DNA binding sites, thus the strange behavior of protein–DNA interaction is not surprising.
2. Rotational correlation function (RCF) and DNA relaxation time
The rotational movement of DNA as elucidated by the phosphate–oxygen (PO) bond vector is important to study the conformational flexibility and propensity of DNA movement. This rotation has been studied in order to find molecular orientation of each molecule by cross-multiplying the paired-atom vectors (i, j). Therefore, RCF was studied with first- and second-order Legendre polynomials and we found that the decay pattern of internal motion was similar in all cases albeit there were differences in the rate of decay (Fig. 2A). RCF of DNAWT decayed slightly and reached a stable value quickly, followed by DNAOCT, DNASOX and free DNAs. The decay in DNAMUT was aberrant and no strong correlation was found. On the other hand, DNANS decayed rapidly in an exponential manner and its value reached zero. Smooth and non-exponential decay was observed in regular systems, whereas anomalous and exponential decay was evident in DNAMUT and DNANS, respectively. | ||
| Fig. 2 The rotational correlation function (RCF) and bend angle of DNA. (A) The internal spin and global rotational movement were characterized using RCF for PO bond vectors of DNA using Legendre polynomial 1 (P1) and 2 (P2). The decay in spin movement was smooth in regular complexes or free DNAs but aberrant in DNAMUT and DNANS complexes. (B) The DNA bend angle according to CURVES+. This angle was calculated between the tangents of successive base pairs of the helix that define the curvature. This is an average of 18 bp, to which SOX2 and OCT4 bind. | ||
DNAWT showed the longest time for relaxation, (i.e., τc = 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 851.2 ps) which may have correlated with a stable interaction with proteins that rigidifies the DNA core, as opposed to DNAMUT (35815.98 ps; where the protein interaction was disrupted), which showed the smallest relaxation time with the same DNA sequence. This finding points to a different kinetic and relaxation mode for the DNA from DNAWT and DNAMUT. Moreover, it was evident that there was a linear relation between the size and nature of the complex with relaxation time in both Legendre polynomials (ESI Table S3†). DNANS has the smallest relaxation time among these complexes: less than a half of DNACRY relaxation time.
851.2 ps) which may have correlated with a stable interaction with proteins that rigidifies the DNA core, as opposed to DNAMUT (35815.98 ps; where the protein interaction was disrupted), which showed the smallest relaxation time with the same DNA sequence. This finding points to a different kinetic and relaxation mode for the DNA from DNAWT and DNAMUT. Moreover, it was evident that there was a linear relation between the size and nature of the complex with relaxation time in both Legendre polynomials (ESI Table S3†). DNANS has the smallest relaxation time among these complexes: less than a half of DNACRY relaxation time.
The SOX2 protein belongs to the helix-turn-helix group of proteins that can bend DNA curvature by up to 50–90° and this bending is essential for this protein's activity;29 therefore, protein-induced DNA bending is necessary to gain an insight into DNA's structural deformation. The axis bend angle of DNA was measured by CURVES+,30 which revealed that DNAWT and DNASOX maintained the bend of ∼70°. DNA curvature in other systems quickly dropped to lower values either due to the absence of SOX2 or due to SOX2-mediated protein–protein interaction (Fig. 2B).
3. Energetics of double-stranded DNA (dsDNA) and protein–DNA interaction
Structural integrity of DNA has been largely attributed to stacking energy among various base pairs that stabilizes the overall conformation; thus, conformational changes in DNA are energetically expensive. Furthermore, the SOX2 protein bends DNA to some extent, which may alter its conformational state. The energetics of DNA conformation also reveal the potential state, namely, whether this configuration is ready to exert biological functions. In order to study this phenomenon, thermodynamic quantities of DNA were determined using MINT.31 MINT calculated these terms by atom-centered point charges where it applied Lennard-Jones and coulombic terms and it has the capability to reproduce ab initio based energy terms within ±-1.5 kcal mol−1. It was found that DNACRY (6.862 ± 1.683 kJ mol−1) has higher mean Coulomb values (ESI Table S4†) but lower mean vdW (van der Waals) energy (−2.93 ± 0.182 kJ mol−1) than the other complexes. Other complexes showed uniform Coulomb and vdW energy values, indicating that all the simulated DNAs preserved their basic interaction efficiency, which can be replicated through molecular mechanics force fields.Another way to measure stability of the complex is to evaluate the binding free energy between a protein and DNA. In this molecular mechanic Poisson Boltzmann surface area (MM-PBSA) approach, various contributions of binding free energies have been calculated by solving Poisson Boltzmann equation for solvation energies and then adjusting the hydrophobic terms empirically. Moreover, vacuum based binding free energy (ΔGvacuum) is the average interaction energy between receptor and protein, and to account for entropy, normal mode analysis could be performed. Interesting results were obtained: SOX2 showed higher binding free energy toward DNA than OCT4 did (Table 1). Proteins in the DNAWT complex showed higher stability in comparison with DNAMUT, and this result can be rationally explained as mutation renders the complex unstable, thus leading to abrogation of the transcriptional activity. The DNANS complex showed positive energy, indicating thermodynamically unfavorable binding of the protein and DNA.
| Energy terms | DNASOX | DNAOCT | DNAWT | DNAMUT | DNANS |
|---|---|---|---|---|---|
| van der Waals | −175.95 (4.09) | −338.26 (6.01) | −360.54 (6.96) | −391.37 (4.89) | −151.6 (3.4) |
| Electrostatic | −9634.8 (33.8) | −19084.8 (90.7) | −20180.4 (71.3) | −19155.18 (32.4) | −9105.5 (47.5) |
| Polar solvation | 9606.3 (24.9) | 19138.4 (74.8) |
20140.26 (76.6) |
18994.48 (27.3) |
9195.5 (38.05) |
| Nonpolar solvation | −109.7 (0.7) | −224.27 (2.4) | −49.13 (0.34) | −261.06 (2.04) | −100.2 (1.9) |
| Dispersion | 225.5 (1.4) | 465.08 (4.8) | 449.8 (13.8) | 510.5 (4.4) | 227.1 (2.06) |
| Emolecular-mechanics | −9810.8 (32.14) | −19423.02 (90.8) | −20540.97 (73.8) | −19546.43 (31.8) | −9257.06 (49.7) |
| ΔGsolvation | 9722.07 (24.6) | 19379.23 (75.6) |
20091.13 (76.4) |
19243.89 (27.7) |
9322.4 (37.96) |
| ΔGbinding | −88.73 (10.4) | −43.8 (18.04) | −360.54 (6.96) | −302.54 (17.8) | 65.3 (20.4) |
A better insight into the configurational space sampled by the DNA during the simulations can be obtained by calculating configurational entropy of DNA atoms along the trajectories. The entropy of DNA was calculated in two ways, the Schlitter formula and quasi-harmonic approximation analysis (Fig. 3A and B) and both method relied on covariance matrix generation after superposition of DNA heavy atoms over the trajectory. The fluctuation from this covariance matrix then used to estimate the entropic values. The entropies in all cases reached a plateau value, but DNASYN and DNACRY showed higher entropies on average. DNA bound to a single protein or a mutated protein and non-specific binding all showed almost identical values. DNA from the DNAWT complex was an exception: it showed the least entropy in both instances. The specific binding can shrink the configurational subspace; this notion is evident in our results and in the literature.32 In DNASOX and DNAOCT, SOX2 and OCT4 also bound to the specific regions, but the absence of a cognate protein could not force the DNA into a lower entropic state.
 | ||
| Fig. 3 DNA entropy. Entropy of the atom-positional fluctuation was calculated, which was derived from the covariance matrices of different complexes. Entropy due to quasi-harmonic approximation (A) and Schlitter's formula (B) over the entire trajectory. | ||
4. Diffusive and dipolar properties and dynamic cross-correlation matrices (DCCMs) are significantly different
DNA mobility in solution was also analyzed for diffusive properties such as the diffusion coefficient that employed the mean square displacement from initial positions of atomic coordinates and then solved the Einstein relation for diffusion coefficient. It was shown that DNAWT has the smallest diffusion coefficient followed by DNAOCT and DNASOX (ESI Table S5†). DNACRY had a diffusion coefficient comparable to that of DNAMUT. DNANS showed an inverse diffusion coefficient; apparently, this is because DNA in non-specific complex is aggregating with time or the proteins bound to DNANS may restrict this random behavior and may negatively influence the diffusion of the bound DNA.Furthermore, DNA is a charged molecule that bends in an asymmetric manner, creating a dipole moment, which is crucial for its activity. Therefore, the dipole moment for the heavy atoms of DNA was calculated to deduce a possible link describing the effect of protein-induced variation on the DNA dipole. Binding of SOX2 could not influence the dipole (DNASYN = 76.6 ± 24.9 D, DNACRY = 90.8 ± 28.1 D and DNASOX = 89.7 ± 23.3 D); however, OCT4 dramatically reduced the dipole (60.6 ± 18 D) and this change was prominent in DNAWT (46.8 ± 22.7 D). DNAMUT (66.5 ± 24.6 D) showed an intermediate dipole moment, while DNANS (33.1 ± 14.1 D) showed the smallest dipole moment.
A DCCM is a good indicator of how various atoms communicate with one another during evolution of coordinates, which demonstrated substantial variations in correlation among DNA atoms (excluding H atoms) in different complexes (Fig. 4). DNA termini showed a strong positive correlation, as expected, a uniform and higher degree of positive correlation was evident in DNANS and DNACRY, respectively. The binding region for SOX2 (which is from atom 38 to atom 178 on the 5′–3′ strand and atoms 589–752 on the 3′–5′ strand) and OCT4 (atoms 243–406 and 812–957 on 5′–3′ and 3′–5′ strands, respectively) favored a positive correlation in DNASOX, DNAOCT, DNAWT and DNANS; in contrast, these regions in DNAMUT lacked any substantial positive correlations. Moreover, the intermediate region between these two binding motifs showed a good positive correlation in DNAWT and DNANS, whereas DNAMUT again lacked any substantial positive correlation. During comparison of SOX2 and OCT4 binding with DNA, it was apparent that the binding of both proteins had distinct effects on correlative behavior of DNA atoms, even the self-correlation among the atoms was restricted. Because of dsDNA, two diagonal lines in the matrices appeared, which also locally influenced the counterparts of each base pair. Dynamically, the binding of proteins influenced not only local but also global DNA movements.
 | ||
| Fig. 4 Dynamic cross-correlation matrices (DCCMs). The interatomic cross-correlation among the heavy atoms of DNA was calculated from the last 100 ns of the trajectory after removal of translational and rotational movements. The legend and color key are shown. | ||
5. SOX2 confers A-like whereas OCT4 confers B-like structural properties onto DNA
The binding of SOX2 to DNA not only bent the DNA but also preferentially configured DNA into a non-standard B-form, whereas OCT4 binding preferentially shaped the bound DNA into the B-form. After the binding, the delicate balance between the A- and B-forms then determines the underlying efficiency and triggering of downstream gene transcription. In particular, this phenomenon was elucidated by means of the coupled distribution of various DNA parameters, and these parameters have been elucidated by CURVES+.30The coupled distribution of roll with twist and slide of all systems is of particular interest; it revealed that the binding of SOX2 to the DNA altered its conformation to a non-standard B-form (or close to A-form), whereas free DNA showed a conformation close to the B-from as other systems did: DNAOCT, DNAMUT, DNAWT and DNANS complexes. The distribution of DNAWT was congested in both cases within a limited space, indicating a strict conformational requirement for the DNA in terms of efficient transcription (Fig. 5). Moreover, there was no correlation in DNAWT, but all the other complexes showed a weak-to-moderate negative correlation in roll/twist parameters, whereas in roll/slide distribution, DNAWT yielded a strong positive correlation, whereas the other systems were moderately-to-weakly positively correlated (ESI Table S6†). Furthermore, coupled distribution between an inclination and helical twist (h-twist) further illuminated the conformational variations in DNA, e.g., with OCT4 or in dual protein-bound complexes. Again, DNASOX showed a higher inclination and lower twist: a characteristic similar to A-form DNA. Only DNAWT showed a weak positive correlation, whereas the other complexes were either uncorrelated or negatively correlated. This finding further clarified the conformational changes of DNA in response to the protein interaction.
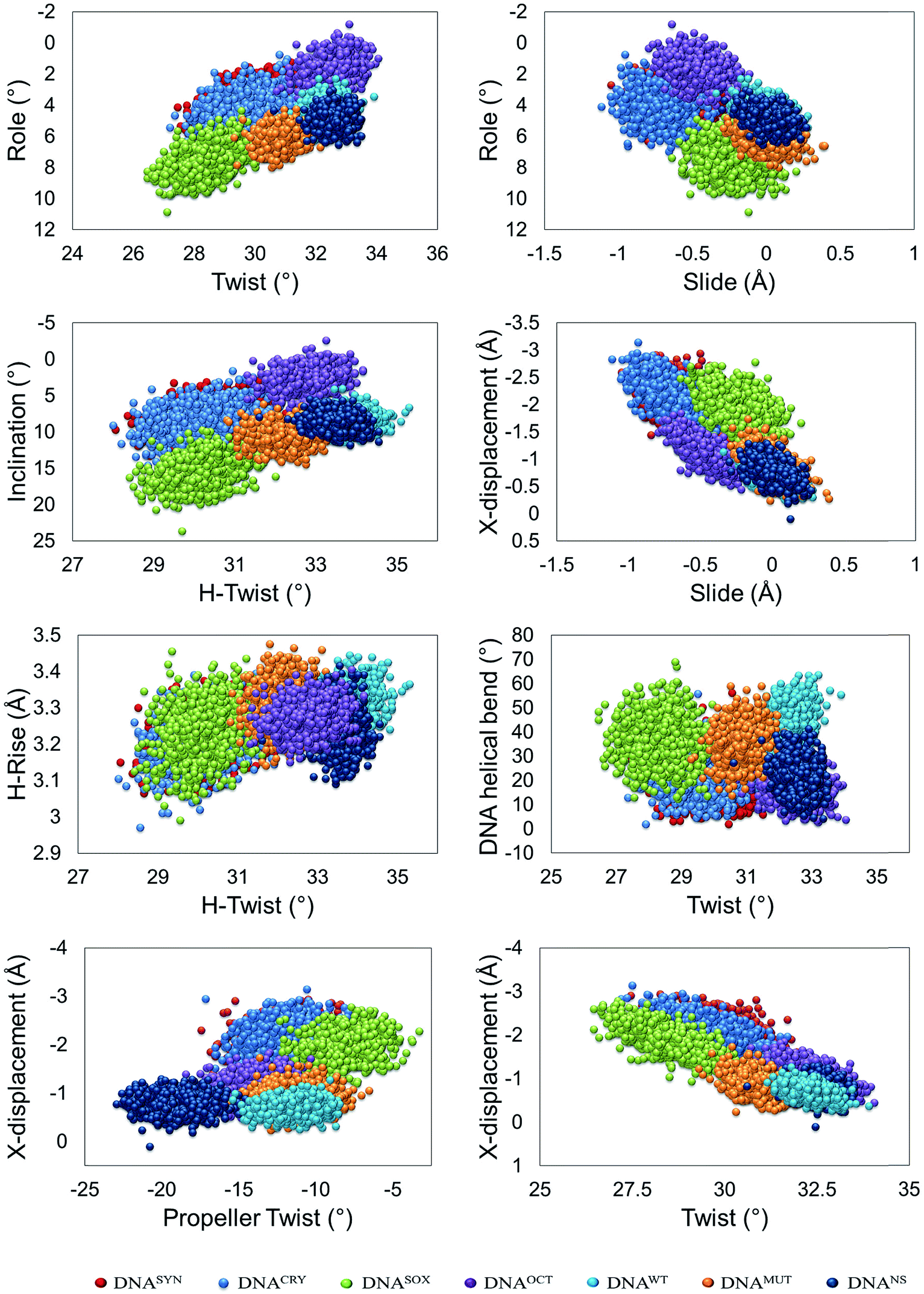 | ||
| Fig. 5 DNA conformational parameters. The distribution of different DNA conformational parameters was calculated by means of CURVES+ from the last 100 ns of each trajectory with equally spaced 1000 frames in each analysis. The distributions were plotted by taking the average of each value from two independent simulations. Only the 18 bp to which SOX2 and OCT4 bound were included in the conformational analysis. Twist refers to the bp stacking geometry, whereas h-twist (helical twist) refers to the helical geometry, and both should be identical in ideal B-DNA conformation, however varies in A-DNA conformation. | ||
Slide and X-displacement coupled distribution is also different, where DNAWT, DNAMUT and DNANS are restricted on a limited space. However, DNASYN and DNACRY are occupying a different space. However, DNASOX and DNAOCT representing a bridge between these two conformational spaces that refer to a critical balance between these two proteins to achieve a stable conformation.
Significant differences were observed in protein-induced DNA bend and twist when conjoined distribution was evaluated. In the DNAWT system, DNA occupied a well-pronounced space as opposed to all the other systems. Moreover, free DNAs and DNASOX were overlapping, with a greater spread of DNASOX in the bend-twist conformational space. Moreover, OCT4 binding restricted the complex in a limited space in contrast to SOX2 binding and it was overlapping with the DNANS region, thus plausibly pointing to the underlying fact that, in the absence of SOX2, OCT4 binding to the specific DNA sites is likely unstable and may behave as a binding to a non-specific region. Moreover, the fact that the SOX2 binding could not achieve a stable space may be related to its small binding domain; in addition, the SOX2 binding triggered a configurational change of DNA to an appropriate conformation that may facilitate the stable binding of OCT4 and yield a productive conformation. Strikingly, DNAWT and DNAMUT had a weak and moderate positive correlation, respectively, but the other systems showed a weak negative correlation between the parameters (Fig. 5).
The coupling distribution of the helical rise and h-twist yielded a similar pattern, i.e., DNAWT, DNAMUT, DNANS and DNAOCT were all in a separated region overlapping one another (Fig. 5), with DNAWT in agreement with the B-form of DNA, in contrast to DNASOX, DNACRY and DNASYN. Other than DNANS, all the complexes showed a weak positive correlation, and the correlation in DNAWT was the weakest. It should be noted that twist (rotation of bp step along z-axis) and helical twist (helical geometry of DNA strand) should be identical in ideal B-DNA conformation, however, it is not the case here that refers to the distorted geometries. Next, X-displacement was plotted against a propeller twist of DNAs that segregated the DNANS complex. Nonetheless, as seen in other distributions, DNAs from DNAOCT, DNAWT and DNAMUT complexes occupied the same region, although the separation was well pronounced, whereas free DNAs and DNASOX occupied an entirely different region. Either no correlation (DNAWT only) or a weak negative correlation was observed (ESI Table S6†). X-Displacement and twist coupling distribution yielded a pattern with different systems occupied a different conformational space, where DNASYN and DNANS showed no correlation, DNAWT showed moderate correlation, while the all other complexes showed the strong positive correlation.
Finally, minor and major grooves of DNA that are critical for its activity have been analyzed in various systems (Fig. 6). SOX2 is a minor-groove-binding protein, which is also known to bend the DNA; therefore, its binding causes the minor groove to expand while the major groove shrinks. This behavior was evident in DNASOX, DNAWT and DNAMUT and in all these cases, SOX2 was binding to its specific binding site. However, the system where SOX2 is binding to non-specific DNA and the systems that do not have SOX2 showed the usual width of minor and major grooves in DNA.
 | ||
| Fig. 6 Groove parameters. The groove parameters were calculated using CURVES+ and plotted against the base pairs. From the last 100 ns, equally spaced 1000 snapshots of DNA were extracted. DNA to which SOX2 and OCT bind was included in analysis. Error bars indicate the standard deviation. | ||
6. Principal component analysis (PCA)
PCA was performed to monitor the protein–DNA cumulative movements. PCA is a standard mathematical tool that employs the orthogonal transformation in order to obtain a set of plausible uncorrelated variables (principal components) from the large data sets. This transformation has been carried out in a way that the largest possible variance can be represented by the first principal component. DNA, either bound or free, showed similar movements (Fig. 7). SOX2 movement was greatly influenced by the presence of OCT4, as in DNASOX, SOX2 was moving outward. OCT4 showed only a counter-clockwise movement in its both domains (POUS and POUHD). | ||
| Fig. 7 Principal component analysis (PCA) of protein–DNA complexes. Backbone atoms from proteins and P, C4′, and C2 atoms of DNA were subjected to PCA analysis as implemented in VMD (using ProDy). The presented figures were drawn on the basis of the first principal component. In all complexes, double-stranded DNA is shown in red, SOX2 in blue, and OCT4 in orange. The moving tendency of protein–DNA complex is represented by green arrows (PC1), with the length of arrows corresponding to the magnitude of deformation; arrowheads show the direction of deformation. | ||
In DNAWT, SOX2 was pointing toward OCT4, with the POUS domain of OCT4 showing restricted movements; however, POUHD conserved its domain movement. The dominant direction of SOX2 in DNAMUT was perpendicular as seen in DNAWT, with POUS showing abrupt movements; however, POUHD movement was influenced by the R113E mutation in the tail part of SOX2. This result is in line with our previously shown RMSF graphs (Fig. 1D), where the mutant complex showed greater fluctuations in POUHD domains. In the DNANS complex, SOX2 was moving away from the dimerization interface, with POUS showing the same pattern as evident in the DNAOCT complex. Nonetheless, the POUHD domain of OCT4 was moving in a clockwise manner (Fig. 7). This is the only instance where the POUHD domain's movement was anomalous. In a non-specific binding, the protein should search for other sequences, which is possible when the protein is flexible and moves along the DNA to find its consensus binding sequence.
The covariance matrices that were calculated by PCA were then cross-multiplied to evaluate the overlap between the matrices, to analyze the most prominent directions of DNA motion, and to determine whether they remain well-defined throughout the trajectory. The normalized value representing the overlap between the matrices was also obtained, which was 1.0 when sampled subspaces were identical and 0 when the sampled subspaces were orthogonal to each other. The root-mean-square inner product (RMSIP) compared the overlap of the first 10 eigenvectors that captured >80% of the magnitude of the overall direction and measured the similarity of the modes that have captured the largest deformational propensity in each structure (Fig. 8). All complexes were compared with DNAWT because it represents the most appropriate conformation. It was noted that the third eigenvector of DNAWT and the first eigenvector of DNACRY and DNASYN shared the same subspace of DNA movement. DNASOX, DNAOCT, DNAMUT and DNANS all shared the passing similarity, but the contribution from the first three eigenvectors from all complexes was substantially different, indicating different directional movement or subspace sampled. In DNAMUT, the overlap of the first eigenvector from both the WT and mutant complexes was significantly smaller. The first eigenvector captured the largest fluctuation of the macromolecule, but in the mutant complex, the dominant direction of the first eigenvector was not overlapping, indicating a loss of cumulative movements of DNA due to the mutation.
 | ||
| Fig. 8 The subspace overlap of covariance matrices. The covariance matrices were calculated for heavy atoms of DNA from the last 100 ns of each system. These matrices were then compared with DNA from the wild-type complex for the first 10 eigenvectors that encompass >80% of the deformation magnitude. Normalized values (1.0 when the sampled subspaces were identical and 0 when the sampled subspaces were orthogonal to each other) are shown in parentheses. | ||
Discussion
The binding of SOX2 and OCT4 to the enhancer region of various genes can influence their expression and modulate cellular physiology during development. Here, we delineated the conformational aspects of the binding of OCT4 and SOX2 to DNA, in particular, we analyzed the case when two proteins bind in an asymmetric manner and their prominent mode of interaction is allosteric over DNA. OCT4 and SOX2 binding interaction over the fibroblast growth factor 4 enhancer was studied due to its well-established pluripotency effect,10 with minimal protein–protein interaction, which can help to determine the precise role of DNA. Eventually, a single mutation severely influenced the complex stability, leading to profound effects during MD simulations.Protein and DNA alone and in complex were stable throughout the simulation, but their residual absolute energies as measured by RMSF were in agreement with the fact that DNAWT has lowest, whereas DNACRY, DNASYN and DNASOX have the highest values, arguably due to tightly bound proteins and absolutely free and/or SOX2 bound DNAs in the latter cases. The protein residues also have lower RMSF in the DNAWT complex than in DNAMUT and DNANS. In the DNAMUT complex, POUHD of OCT4 showed greater fluctuations, which may also indicate a protein–protein interaction's being perpetuated among the protein domains.8 Substitution of one amino acid residue can considerably influence the protein's conformational flexibility (Fig. 1C and D).33 This may also indicate the delicate balance of interactions among SOX2, OCT4 and DNA to tightly monitor the induction of target genes that is a stringent requirement of this process, and is also in line to previous studies that highlighted the protein–protein interaction through SOX2's tail part in various complexes.6,8 The number of H-bonds within dsDNA is within the potential number of H-bonds between the bases and indicates the ability of the force field to reproduce the natural phenomenon. Moreover, these analyses add an additional layer of confidence over the results.
Conformationally, DNA is not limited to the standard A-, B-, or Z-forms. A growing amount of evidence suggests that protein binding causes DNA to assume an A-like or an A–B intermediate conformation in terms of DNA parameters such as roll and twist.34,35 Moreover, there are >15 parameters that can be measured using the standard DNA parametric algorithms, such as CURVES+,30 but a large number of parameters are overlapping and lack the decisive power for DNA classification. On the other hand, the slide and X-displacement hold this potential; for instance, it is widely accepted that a lower and higher slide value (−0.8 Å) are characteristics of A- or B-form DNA, respectively.36 In line to this, we observed overlapping parameters for the standard A and B conformations. In general, SOX2 binding caused DNA to acquire an A-like conformation, while OCT4 alone binding is unable to alter the canonical shape of the DNA. It is arguable whether OCT4 sparsely bound to DNA at the first place, so OCT4 binding could not influence the DNA conformation significantly. In contrast, when both proteins bind to DNA, the resultant conformation is either in between A and B forms, or close to B form. However, free DNAs are more like the B-form and strikingly overlap each other that indicates the convergence of simulations irrespective of the starting conformation. Moreover, SOX2 bent DNA by up to 50–90° (Fig. 2B and other studies37,38); this phenomenon may also switch its conformation from a stranded B-form to non-stranded B- or A-like conformations.
In addition to experimental approaches, theoretical studies have also pointed to the conformational transition in DNA from B- to non-B and/or A-like conformation upon protein binding or an intermediate stage between A and B during DNA distortion.39–41 For instance, the binding region of TATA box-binding protein in DNA converged to an A-like conformation in longer simulations irrespective of starting conformations;42 in Escherichia coli, cyclic AMP receptor protein can induce A-form upon binding when its recognition motif has an 8-bp spacer,43 which may be necessary to shorten the motif. A polymerase-induced A conformation of DNA has also been detected during HIV reverse transcriptase binding to DNA44 that eliminates error during replication.45 Our study also predicts that DNAOCT plausibly assumes the B-form, DNASOX preferably adopts an A-like form and DNA in the DNAWT complex attempts to adopt an intermediate conformation between the A- and B-forms (Fig. 5 and ESI Table S6†). This finding may explain why for regulation of a complex process like stem cell generation, a very specific conformational requirement has been imposed during evolution to ensure precise control over the process.
SOX2 and OCT4 can bind to DNA in two predominant modes, with a 0- and 3-bp gap within their binding sites. Both motifs organizations produce different binding interfaces that result in different intensity of the expression of target genes.46,47 Nonetheless, the binding-motif gap is evident in other complexes like SOX2/OCT4 in the Zfp206 enhancer,48 SOX2 and PAX6 (paired box genes 6) interaction,49 SOX2 and brain-specific homeobox/POU domain protein 2 (BRN2)50 and SOX1/2/3 and the POU family.51 In the present study, we used the available crystal structure data, and the results can be extrapolated to other similar complexes, where SOX2 plays a vital role in transcription. Moreover, there are numerous complexes, for instance, dyskerin52 and trimeric xeroderma pigmentosum complementation group C (XPC)-nucleotide excision repair complex53 that used to modulate SOX2/OCT4 activities over DNA. The involvement of many proteins in regulation and monitoring of the activity of this SOX2/OCT4 pair points to the importance of their regulated behavior during embryogenesis and development.
DNA exhibits dipole moment due to its inherent asymmetric bending nature, but when SOX2 binds to DNA, it also bends the DNA even though it cannot influence the dipole moment. Whereas, OCT4 is a bulky protein that has influenced the dipole moment. In order to confirm the results, simulations have been conducted again for 100 ns in each case, but the similar results were observed. Besides, AMBER force field is good at reproducing the structural features, but they are lagging behind with respect to the physical parameters.54 Moreover, the number of water molecules around the DNA is different in different systems that may also influence the dipole moment. Nevertheless, the choice of a force field is a compromise among certain structural and physical properties.
The interactions of biomolecules and ligands in solution depend not only on their binding affinity but also on their translational diffusion coefficients. DNAWT has the smallest diffusion coefficient with the longest relaxation time, which indicates a lower mobility and a rigid molecular configuration (ESI Table S5†). The longer relaxation time of a polymer like DNA is fundamentally important because it defines the natural time constant of the molecule and characterizes how rapidly the polymer can react in response to an imposed flow. DNAs from mutant and non-specific complexes have anomalously greater diffusion coefficients (by a factor of ∼30 and ∼300, respectively), but the values of their relaxation time are shorter than that of DNAWT. The flexibility of DNA in mutant and non-specific complexes allows for fast interaction with different proteins during the search for specific binding between the protein and DNA. Experimental and theoretical studies estimated the diffusion coefficient of DNA-binding proteins that are in close proximity;32,55 however, in the present study, unconstrained atomic simulations were conducted to study the DNA behavior. Furthermore, rotation-coupled translational diffusion is a popular concept in terms of protein movement along DNA that is also influenced by polarity of amino acid residues and pH of the solution.56,57 The DNA in the mutant complex was not encountering the same level of hindrance from the bound protein, whereas in non-specific complex, DNA was behaving randomly; this finding corroborates a non-specific protein–DNA interaction.57 For the other systems, relaxation time gradually increased from free DNA to bound DNA, while an inverse pattern was observed for the diffusion constant.
Binding energies were calculated by the method of molecular mechanics with Poisson–Boltzmann surface area (MM/PBSA), though DNA is not comparable to ligand in size, electrostatic density is higher and the ligand (dsDNA) is a double-stranded molecule.47 Despite these flaws, qualitative results can be obtained by MM/PBSA, with a uniform dielectric constant to make the comparison straightforward; however, the higher uncertainty in the computed binding free energies may be attributed to the algorithm that was used to compute free energy.58 These uncertainties are an important factor for ranking of DNA sequences by binding energies, but ranking was not the purpose of this study. In short, the DNAWT complex has the highest binding free energy, followed by DNAMUT, DNASOX and DNAOCT (Table 1). For complexes DNAWT, DNAMUT and DNANS, if entropic contribution could be ignored, the values showed stability of WT protein to their cognate DNA complex. Moreover, a superficial estimate can be made between DNASOX and DNAOCT. SOX2 has fewer amino acid residues (79) than OCT4 does (152), but DNASOX showed greater values than DNAOCT did. Even if an equal entropy contribution can be assumed, the SOX2–DNA complex is more stable.
The direction of SOX2 motion is significantly influenced by OCT4, whereas OCT4's motion direction is also perturbed by SOX2 but to a slightly lesser extent because OCT4 has two domains that wrap around the DNA. In DNAWT complex, their cumulative movement was substantially different from that in the DNAMUT and DNANS complexes (Fig. 7). Subspace sampling revealed significant directional discrepancies in DNA atoms; these data reinforce the concept that protein binding changes magnitude and alters the direction of bound DNA (Fig. 8).59 Further theoretical and practical studies are underway to prove and design various strategies to manipulate this complex.
Besides the theoretical studies such as molecular modeling and molecular dynamics simulations, X-ray crystallography and nuclear magnetic resonance (NMR) can be employed to decipher the structural details; however, these techniques are laborious and have limitations. Atomic force microscopy is the latest technique that holds potential to unravel the structural details without modifying the biological material. Moreover, single molecule force microscopy can be also a good tool to study the protein–DNA interaction. In conclusion, despite DNA-mediated allostery, DNA shape alteration that is mediated by protein–protein interaction plays a crucial role in complex organization, and the relative participation of each molecule, where SOX2 has a greater influence on DNA shape then OCT4 does. Nonetheless, an intense interplay between SOX2 and OCT4 determines the complex's biological efficiency. Moreover, SOX2-mediated DNA bend, SOX2/OCT4-mediated switch in DNA shape and a balance between these DNA conformations are critical factors that substantially influence the complex's biological efficiency. Further, there can be various ways to either disrupt or stabilize their interaction, such as hindering the binding of SOX2 to DNA, or altering the DNA structure despite their binding using peptide-nucleic acids. However, the manipulation of this ternary complex is feasible after in depth study of the molecular architecture, the respective role of each component in complex formation and atomic level details of their interaction. Therefore, this study will be the starting point of a broad spectrum analysis that can provide a reference to target a multitude of challenges in regenerative medicine and cancer therapy.
Experimental procedure
1. Initial structures
The initial coordinates of OCT1/SOX2/DNA were retrieved from the RCSB Protein Data Bank (PDB). To date, crystal structure of the OCT4/SOX2/DNA complex has not been reported; therefore, we modeled the complex with a few modifications in the existing crystal structure of OCT1/SOX2/DNA (1GT0)10 and 3L1P.27 Coordinates of the missing amino acids were built using homology modeling through MODLLER.60 The coordinates of completed OCT4 were used to replace OCT1 translationally in the complex thus constituting the whole complex.2. Structure manipulation and complexes generation
The mutated complex was generated in the Chimera v1.9 software by replacing the arginine amino acid residue (of SOX2 at the 113th position) with the most appropriate glutamate rotamer, followed by energy minimization. The synthetic DNA was created using 3DDART61 after providing a single sequence with default parameters as per B-DNA conformation. Other complexes were generated by deleting one or the other or both proteins from the whole complex.Seven systems were generated: DNA was extracted from the crystal structure 1GT0 (DNACRY); with the same sequence, a perfect B-DNA was created using 3DAART (DNASYN), crystal structure with DNA and SOX2 (DNASOX) and of DNA and OCT4 (DNAOCT) were also set up; the whole crystal structure with wild type SOX2 (DNAWT), mutated SOX2 (R113E; DNAMUT) and finally a non-specific complex that was generated by mutating the DNA to a non-specific sequence (DNANS). DNASYN and DNACRY represent DNA alone (without any protein) to simulate as a reference and to evaluate DNA movement.
3. Molecular dynamics simulations
The detailed method of MD simulations is described in our previous publications.62,63 Briefly, for these simulations, GROMACSv5.0.7 (ref. 64) was used. In each case, a dodecahedron box filled with TIP3P water model65 was generated. For atomic representation, the AMBER99SB-ILDN66 force field parameters were selected. The box dimension was properly adjusted to account for minimum image conventions and periodic boundary conditions were applied in all directions to mimic the infinite system. The Particle mesh Ewald approach was employed for long-range electrostatics67 using a 10 Å cut-off distance for both electrostatic and van der Waals interactions and the dispersion correction was applied. Bond lengths were constrained using LINCS algorithm68 that allowed for a 2 fs time step for all simulations. Before the simulation, all the systems were neutralized and 100 mM NaCl was added to mimic physiological conditions. The steepest descent and/or conjugate gradient minimization (with maximum tolerance of 100 kJ [mol nm]−1) were performed to remove any unfavorable interactions. Each system was prepared for the production simulation in accordance with two-step equilibration. During the first stage, the systems were simulated in a constant volume (NVT) ensemble to achieve 310 K by the V-rescale method69 for 500 ps. The equilibrated structures from the NVT ensemble were subjected to constant pressure (NPT) equilibration (500 ps) using the Parrinello–Rahman barostat70 under isotropic pressure of 1.0 bar. To avoid configuration changes during equilibration, position restraints were applied to all solute atoms. Production MD simulations were performed for 250 ns in the absence of any restraints and the coordinates have been saved at every 5 ps interval. During the data collection period, the V-rescale thermostat and Parrinello–Rahman barostat were used to maintain the temperature and pressure at 310 K and 1 bar, respectively.Simulations were repeated with the same parameters with a slight difference because the starting structures were extracted approximately after 150 ns of the previous trajectory. This way, the equilibration period could be ignored during analysis in the repeated simulation. All complexes were then energy minimized, equilibrated for temperature and pressure, and simulated for additional 100 ns.
4. Dynamics cross-correlation matrices (DCCM)
DCCM have been calculated using Bio3D package71 of the R-software v3.2.3. For DCCM calculation, from last 100 ns of simulation, 1000 equidistance snapshots of DNA heavy atoms have been subjected for calculations.5. Energetics and entropy calculations
The various forms of energies have been calculated through different methods. The stacking energies between base pairs have been analyzed using MINT31 and represented as the average for each system. The binding free energies between DNA and proteins have been studied by AMBER Tools. Finally, the conformational entropy has been extracted from the covariance matrices of positional fluctuation of each system after removing translational and rotational movements.6. DNA parameters
To study the DNA parameters, CURVES+30 has been used. Equally spaced snapshots of DNA have been extracted from last 100 ns of each trajectory and various parameters including axis related base pairs, intra- and inter-base pairs have been calculated as per default parameters. Average values from two independent simulation runs have been plotted.7. Principal component analysis (PCA)
PCA was calculated through GROMACSv5.0.7 implemented tools. In this, the coordinates from trajectory were fitted to reference structure, calculated the non-mass-weighted covariance matrix, which was later diagonalized. The diagonalized matrices were then cross-multiplied to monitor the overlap and plausible movement direction. Further, PCA was also calculated and figures were drawn by means of Visual Molecular Dynamics (VMD).728. Data analysis
For most of the analyses, built-in tools of GROMACSv5.0.7 were used and the analyses were performed during the last 100 ns unless otherwise stated. The figures were drawn by Xmgrace, PyMol (http://www.pymol.org) and VMD.Competing financial interests
The authors declare that they have no conflicts of interest with the contents of this article.Author contributions
MAA and SC designed the study. MAA, DY, and MS performed the experiments and analyzed the data. SC contributed materials. MAA, DY, MS, and SC wrote the manuscript.Abbreviations
| BRN | Brain-specific homeobox/POU domain protein 2 |
| DCCM | Dynamic cross-correlation matrix |
| HMG | High mobility group |
| iPSCs | Induced pluripotent stem cells |
| MDS | Molecular dynamic simulations |
| MM/PBSA | Molecular mechanics Poisson–Boltzmann surface area |
| OCT4 | Octamer-binding transcription factor 4 |
| PAX | Paired box genes |
| PCA | Principal component analysis |
| POU | Pit-Oct-Unc family |
| RCF | Rotational correlation function |
| RMSD | Root mean square deviation |
| RMSF | Root-mean square fluctuation |
| RMSIP | Root mean square inner product |
| SOX2 | Sex determining region Y-box 2 |
| WT | Wild type |
| XPC | Xeroderma pigmentosum complementation group C |
Acknowledgements
This work was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (HI14C1992) and by the Mid-Career Researcher Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (NRF-2015R1A2A2A09001059). This work was also partially supported by a grant from the Priority Research Centers Program (NRF 2012-0006687). MAA is grateful to Usman Anwar and Rizwan Qamar for their help with the analysis.References
- A. B. Georges, B. A. Benayoun, S. Caburet and R. A. Veitia, FASEB J., 2010, 24, 346–356 CrossRef CAS PubMed.
- K. M. Lelli, M. Slattery and R. S. Mann, Annu. Rev. Genet., 2012, 46, 43–68 CrossRef CAS PubMed.
- M. Slattery, T. Riley, P. Liu, N. Abe, P. Gomez-Alcala, I. Dror, T. Zhou, R. Rohs, B. Honig, H. J. Bussemaker and R. S. Mann, Cell, 2011, 147, 1270–1282 CrossRef CAS PubMed.
- S. Kim, E. Brostromer, D. Xing, J. Jin, S. Chong, H. Ge, S. Wang, C. Gu, L. Yang, Y. Q. Gao, X. D. Su, Y. Sun and X. S. Xie, Science, 2013, 339, 816–819 CrossRef CAS PubMed.
- D. Panne, Curr. Opin. Struct. Biol., 2008, 18, 236–242 CrossRef CAS PubMed.
- K. Narasimhan, S. Pillay, Y. H. Huang, S. Jayabal, B. Udayasuryan, V. Veerapandian, P. Kolatkar, V. Cojocaru, K. Pervushin and R. Jauch, Nucleic Acids Res., 2015, 43, 1513–1528 CrossRef CAS PubMed.
- D. Panne, T. Maniatis and S. C. Harrison, Cell, 2007, 129, 1111–1123 CrossRef CAS PubMed.
- F. Merino, B. Bouvier and V. Cojocaru, PLoS Comput. Biol., 2015, 11, e1004287 Search PubMed.
- Y. Takayama and G. M. Clore, J. Biol. Chem., 2012, 287, 26962–26970 CrossRef CAS PubMed.
- A. Remenyi, K. Lins, L. J. Nissen, R. Reinbold, H. R. Scholer and M. Wilmanns, Genes Dev., 2003, 17, 2048–2059 CrossRef CAS PubMed.
- D. Tantin, Development, 2013, 140, 2857–2866 CrossRef CAS PubMed.
- S. Jerabek, F. Merino, H. R. Scholer and V. Cojocaru, Biochim. Biophys. Acta, 2014, 1839, 138–154 CrossRef CAS PubMed.
- J. Chen, Z. Zhang, L. Li, B. C. Chen, A. Revyakin, B. Hajj, W. Legant, M. Dahan, T. Lionnet, E. Betzig, R. Tjian and Z. Liu, Cell, 2014, 156, 1274–1285 CrossRef CAS PubMed.
- X. Chen, H. Xu, P. Yuan, F. Fang, M. Huss, V. B. Vega, E. Wong, Y. L. Orlov, W. Zhang, J. Jiang, Y. H. Loh, H. C. Yeo, Z. X. Yeo, V. Narang, K. R. Govindarajan, B. Leong, A. Shahab, Y. Ruan, G. Bourque, W. K. Sung, N. D. Clarke, C. L. Wei and H. H. Ng, Cell, 2008, 133, 1106–1117 CrossRef CAS PubMed.
- M. van de Wetering, M. Oosterwegel, K. van Norren and H. Clevers, EMBO J., 1993, 12, 3847–3854 CAS.
- H. Kondoh and Y. Kamachi, Int. J. Biochem. Cell Biol., 2010, 42, 391–399 CrossRef CAS PubMed.
- C. K. Ng, N. X. Li, S. Chee, S. Prabhakar, P. R. Kolatkar and R. Jauch, Nucleic Acids Res., 2012, 40, 4933–4941 CrossRef CAS PubMed.
- C. S. Lam, T. K. Mistri, Y. H. Foo, T. Sudhaharan, H. T. Gan, D. Rodda, L. H. Lim, C. Chou, P. Robson, T. Wohland and S. Ahmed, Biochem. J., 2012, 448, 21–33 CrossRef CAS PubMed.
- A. Soufi, G. Donahue and K. S. Zaret, Cell, 2012, 151, 994–1004 CrossRef CAS PubMed.
- D. C. Ambrosetti, H. R. Scholer, L. Dailey and C. Basilico, J. Biol. Chem., 2000, 275, 23387–23397 CrossRef CAS PubMed.
- M. Nishimoto, A. Fukushima, A. Okuda and M. Muramatsu, Mol. Cell. Biol., 1999, 19, 5453–5465 CrossRef CAS PubMed.
- R. Jauch, I. Aksoy, A. P. Hutchins, C. K. Ng, X. F. Tian, J. Chen, P. Palasingam, P. Robson, L. W. Stanton and P. R. Kolatkar, Stem Cells, 2011, 29, 940–951 CrossRef CAS PubMed.
- S. Stefanovic, N. Abboud, S. Desilets, D. Nury, C. Cowan and M. Puceat, J. Cell Biol., 2009, 186, 665–673 CAS.
- S. Boumahdi, G. Driessens, G. Lapouge, S. Rorive, D. Nassar, M. Le Mercier, B. Delatte, A. Caauwe, S. Lenglez, E. Nkusi, S. Brohee, I. Salmon, C. Dubois, V. del Marmol, F. Fuks, B. Beck and C. Blanpain, Nature, 2014, 511, 246–250 CrossRef CAS PubMed.
- T. Tian, Y. Zhang, S. Wang, J. Zhou and S. Xu, J. Biomed. Res., 2012, 26, 336–345 CrossRef CAS PubMed.
- D. C. Williams Jr, M. Cai and G. M. Clore, J. Biol. Chem., 2004, 279, 1449–1457 CrossRef PubMed.
- D. Esch, J. Vahokoski, M. R. Groves, V. Pogenberg, V. Cojocaru, H. Vom Bruch, D. Han, H. C. Drexler, M. J. Arauzo-Bravo, C. K. Ng, R. Jauch, M. Wilmanns and H. R. Scholer, Nat. Cell Biol., 2013, 15, 295–301 CrossRef CAS PubMed.
- A. Luzar and D. Chandler, Nature, 1996, 379, 55–57 CrossRef CAS.
- P. Scaffidi and M. E. Bianchi, J. Biol. Chem., 2001, 276, 47296–47302 CrossRef CAS PubMed.
- C. Blanchet, M. Pasi, K. Zakrzewska and R. Lavery, Nucleic Acids Res., 2011, 39, W68–W73 CrossRef CAS PubMed.
- A. Gorska, M. Jasinski and J. Trylska, Nucleic Acids Res., 2015, 43, e114 CrossRef PubMed.
- S. Furini, P. Barbini and C. Domene, Nucleic Acids Res., 2013, 41, 3963–3972 CrossRef CAS PubMed.
- C. G. P. Doss, B. Rajith, N. Garwasis, P. R. Mathew, A. S. Raju, K. Apoorva, D. William, N. R. Sadhana, T. Himani and I. P. Dike, Appl. Transl. Genomics, 2012, 1, 37–43 CrossRef CAS.
- W. K. Olson, A. A. Gorin, X. J. Lu, L. M. Hock and V. B. Zhurkin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11163–11168 CrossRef CAS.
- S. Jones, P. van Heyningen, H. M. Berman and J. M. Thornton, J. Mol. Biol., 1999, 287, 877–896 CrossRef CAS PubMed.
- C. R. Calladine and H. R. Drew, J. Mol. Biol., 1984, 178, 773–782 CrossRef CAS PubMed.
- Y. Kamachi, M. Uchikawa and H. Kondoh, Trends Genet., 2000, 16, 182–187 CrossRef CAS PubMed.
- L. Dailey and C. Basilico, J. Cell. Physiol., 2001, 186, 315–328 CrossRef CAS PubMed.
- L. Nekludova and C. O. Pabo, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6948–6952 CrossRef CAS.
- Y. Timsit, J. Mol. Biol., 1999, 293, 835–853 CrossRef CAS PubMed.
- X. J. Lu, Z. Shakked and W. K. Olson, J. Mol. Biol., 2000, 300, 819–840 CrossRef CAS PubMed.
- D. Flatters, M. Young, D. L. Beveridge and R. Lavery, J. Biomol. Struct. Dyn., 1997, 14, 757–765 CAS.
- V. I. Ivanov, L. E. Minchenkova, B. K. Chernov, P. McPhie, S. Ryu, S. Garges, A. M. Barber, V. B. Zhurkin and S. Adhya, J. Mol. Biol., 1995, 245, 228–240 CrossRef CAS PubMed.
- A. Jacobo-Molina, J. Ding, R. G. Nanni, A. D. Clark Jr, X. Lu, C. Tantillo, R. L. Williams, G. Kamer, A. L. Ferris and P. Clark, et al., Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6320–6324 CrossRef CAS.
- J. R. Kiefer, C. Mao, J. C. Braman and L. S. Beese, Nature, 1998, 391, 304–307 CrossRef CAS PubMed.
- N. Tapia, C. MacCarthy, D. Esch, A. Gabriele Marthaler, U. Tiemann, M. J. Arauzo-Bravo, R. Jauch, V. Cojocaru and H. R. Scholer, Sci. Rep., 2015, 5, 13533 CrossRef CAS PubMed.
- D. Yesudhas, M. A. Anwar, S. Panneerselvam, P. Durai, M. Shah and S. Choi, PLoS One, 2016, 11, e0147240 Search PubMed.
- Z. X. Wang, C. H. Teh, J. L. Kueh, T. Lufkin, P. Robson and L. W. Stanton, J. Biol. Chem., 2007, 282, 12822–12830 CrossRef CAS PubMed.
- Y. Kamachi, M. Uchikawa, A. Tanouchi, R. Sekido and H. Kondoh, Genes Dev., 2001, 15, 1272–1286 CrossRef CAS PubMed.
- M. A. Lodato, C. W. Ng, J. A. Wamstad, A. W. Cheng, K. K. Thai, E. Fraenkel, R. Jaenisch and L. A. Boyer, PLoS Genet., 2013, 9, e1003288 CAS.
- S. Tanaka, Y. Kamachi, A. Tanouchi, H. Hamada, N. Jing and H. Kondoh, Mol. Cell. Biol., 2004, 24, 8834–8846 CrossRef CAS PubMed.
- Y. W. Fong, J. J. Ho, C. Inouye and R. Tjian, eLife, 2014, 3, e03573 Search PubMed.
- Y. W. Fong, C. Inouye, T. Yamaguchi, C. Cattoglio, I. Grubisic and R. Tjian, Cell, 2011, 147, 120–131 CrossRef CAS PubMed.
- O. Guvench and A. D. MacKerell Jr, Methods Mol. Biol., 2008, 443, 63–88 CAS.
- S. Furini, C. Domene and S. Cavalcanti, J. Phys. Chem. B, 2010, 114, 2238–2245 CrossRef CAS PubMed.
- P. C. Blainey, G. Luo, S. C. Kou, W. F. Mangel, G. L. Verdine, B. Bagchi and X. S. Xie, Nat. Struct. Mol. Biol., 2009, 16, 1224–1229 CAS.
- Y. Takayama and G. M. Clore, J. Biol. Chem., 2012, 287, 14349–14363 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- J. D. Faraldo-Gomez, L. R. Forrest, M. Baaden, P. J. Bond, C. Domene, G. Patargias, J. Cuthbertson and M. S. Sansom, Proteins, 2004, 57, 783–791 CrossRef CAS PubMed.
- N. Eswar, B. Webb, M. A. Marti-Renom, M. S. Madhusudhan, D. Eramian, M. Y. Shen, U. Pieper and A. Sali, Current Protocols in Protein Science, 2007, 50, 2.9.1–2.9.31 Search PubMed.
- M. van Dijk and A. M. Bonvin, Nucleic Acids Res., 2009, 37, W235–W239 CrossRef CAS PubMed.
- M. A. Anwar, S. Panneerselvam, M. Shah and S. Choi, Sci. Rep., 2015, 5, 7657 CrossRef CAS PubMed.
- M. Shah, M. A. Anwar, S. Park, S. S. Jafri and S. Choi, Sci. Rep., 2015, 5, 13446 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. J. Grant, A. P. Rodrigues, K. M. ElSawy, J. A. McCammon and L. S. Caves, Bioinformatics, 2006, 22, 2695–2696 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15176k |
| This journal is © The Royal Society of Chemistry 2016 |