
1,4-Dihydroxyanthraquinone–copper(II) supported on superparamagnetic Fe3O4@SiO2: an efficient catalyst for N-arylation of nitrogen heterocycles and alkylamines with aryl halides and click synthesis of 1-aryl-1,2,3-triazole derivatives†
Saeed Zahmatkesh*a,
Mohsen Esmaeilpour*b and
Jaber Javidicd
aDepartment of Science, Payame Noor University (PNU), 19395-4697, Tehran, Islamic Republic of Iran. E-mail: zahmatkesh1355@yahoo.com; Fax: +98 7112286008; Tel: +98 7116137738
bChemistry Department, College of Science, Shiraz University, Shiraz, Iran. E-mail: m1250m551085@yahoo.com
cDepartment of Pharmaceutics, School of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran
dStudents Research Committee, School of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran
First published on 12th September 2016
Abstract
1,4-Dihydroxyanthraquinone–copper(II) supported on a superparamagnetic Fe3O4@SiO2 catalyst was employed for the N-arylation of nitrogen heterocycles and alkylamines with aryl halides to afford the corresponding coupled products in good to excellent yields without using external ligands or additives as promoters. Also, we have reported this recyclable catalytic system for efficient synthesis of 1-aryl-1,2,3-triazole derivatives in excellent yields. The desired triazoles were obtained from the reaction of the corresponding aryl boronic acid derivatives, alkyne, NaN3, and 0.5 mol% catalyst in water/acetonitrile as the solvent at room temperature without the additional use of an external reducing agent. These methods show notable advantages such as the heterogeneous nature of the catalyst, low catalyst loading, easy preparation, excellent yields, short reaction times and simplicity of operation. Also, the catalyst can be separated from the reaction mixture by applying a permanent magnet externally and can be reused in six consecutive reaction cycles without significant loss of activity.
1. Introduction
Currently the design of heterogeneous metal catalysts that preserve stability and high metal dispersion during the catalytic reactions is of great importance.1–4 These heterogenized catalysts can be easily separated from the reaction mixtures, but, homogeneous catalysts show higher catalytic activities than their heterogeneous counterparts because of their solubility in reaction media, which increases catalytic site accessibility for the substrate.5–7 However, recycling homogeneous catalysts is often tedious and also it is complicated and costly to recycle homogeneous catalysts, especially when noble and toxic metal complexes are used.8,9 To solve these problems, the particles are often immobilized on various inorganic and organic supports, such as metal oxides, mesoporous silica, polymers, carbon materials, ionic liquids, and so on.10–14The use of magnetic materials as immobilized supports for heterogeneous catalysis has attracted a great deal of attention because of the merits of magnetic separation.15–17 Among transition metal nanoparticle catalysts, Fe3O4 nanoparticles have recently received a lot of attention as excellent supports because of their unique properties including low toxicity, large surface-to-volume ratio, biocompatibility, superparamagnetism and their potential applications in various fields.18,19 Fe3O4 magnetic nanoparticles have been used in wide range of disciplines, including magnetic resonance imaging, magnetic data storage, targeted gene therapy, magnetic fluids, drug delivery systems, biotechnology/biomedicine, hyperthermia treatment of cancer cells, biosensors, environmental remediation, ion exchange separation, detoxification of biological fluids and catalysis.20–28 Also, magnetic separation could be regarded as an attractive alternative to centrifugation or alternative as it prevents the loss of catalyst and enhances its reusability.29
However, pure MNPs such as Fe3O4 nanoparticles trend to aggregate due to their nanoscale and poorly dispersed in aqueous medium due to their hydrophobic surface.30 This problem can be solved by coating of MNPs with inorganic shells or hydrophilic polymers. Among these, coating with a silica layer as the stabilizer, which prevents direct contact between the nanoparticles has attracted great attention in recent years. Furthermore, the abound hydroxyl groups on the surface of silica layer provide the opportunity to conjugate various function molecules for many special applications.31,32
The formation of carbon–nitrogen bonds via cross-coupling reactions represents a powerful means for the preparation of various compounds important in pharmaceutical, biological and material interest.33,34 Copper-catalyzed Ullmann-type reactions are traditional methods to assemble these compounds.35,36 Nucleophilic aromatic substitution with aryl halides can be mediated by copper or palladium catalysts.37 The high cost and toxicity of palladium catalysts restrict their use on the industrial applications. Thus, researchers have turned their attention toward the use of more efficient metals, less expensive, and less toxic to replace palladium. The lower cost of copper-based catalytic systems makes them particularly attractive for large-scale industrial applications.38 During the past years, several reports describing copper-catalysed methods have appeared in the literature.39,40 However, these copper-mediated methods often suffer from limitations such as drastic reaction conditions, lack of generality, moderate yields, high cost and requirement for stoichiometric or greater amounts of copper catalyst. Therefore, the development of a new catalytic system to overcome these shortcomings and fulfill the criteria of a mild, efficient protocol for C–N bond formation is still desirable and is in demand.
1,2,3-Triazoles are an important class of nitrogen heterocyclic compounds which have biological activities and also wide applications in medicinal chemistry, material science and synthesis.41,42 The main method for the synthesis of 1,2,3-triazoles is the Huisgen 1,3-dipolar cycloaddition reaction of azides with alkynes. This cycloaddition reaction has become the model for click reactions.43 However, the disadvantage of Huisgen's reaction was the production of a mixture of 1,4 and 1,5-disubstituted products.43 Consequently, finding new protocols for stereoselective triazole synthesis is of interest.
In 2002, Rostovtsev et al. reported the efficient synthesis of 1,2,3-triazoles form the CuI-catalyzed azide/alkyne cycloaddition (CuAAC). Their methodology is completely stereoselective which delivered 1,4-disubstituted triazoles under mild reaction conditions.44 Since then, the “Click Chemistry” reaction between azide and alkyne (CuAAC) has gained much attention and many copper catalysts were reported for this transformation specially the heterogeneous ones.45 Therefore, variety of supports such as zeolites,46 activated charcoal,47 silica48 and amine functionalized polymers49 were used to immobilize CuI onto their surface. The problem frequently encountered with applying these catalysts is the oxidation tendency of CuI to CuII or disproportion to Cu0 and CuII which reduces the catalytic activity.50 To overcome this matter, other groups have applied CuII in the presence of reducing agents or ligands51 and the most general one is the use of CuSO4/sodium ascorbate in aqueous media.52 In addition, variety of immobilized CuII were reported for 1,2,3-triazoles synthesis.53 Recently, hydroxyapatite-supported CuII (ref. 54) or CuII-hydrotalcite55 were reported for formation of 1,2,3-triazoles in the absence of base or reducing agents. They have reported that NaN3 can reduce CuII to CuI beyond being an azidonation reagent. Many of the reported synthetic protocols for the synthesis of triazole derivatives suffer from one or more disadvantages such as the harsh reaction conditions, long reaction times, low yields, tedious workup of the reaction mixture and difficulty in separation and recovery of the catalyst. Thus, the development of an alternate milder and cleaner procedure, which surpasses those limitations, is very much relevant for the synthesis of 1-aryl-1,2,3-triazole derivatives.
Therefore, in continuation of our studies and to promote the methods of new synthetic using recyclable catalysts,56 we focused our attention on a simple and efficient method for N-arylation of nitrogen heterocycles and alkylamines with aryl halides and click synthesis of 1-aryl-1,2,3-triazole derivatives in the presence of 1,4-dihydroxyanthraquinone–copper(II) immobilized on superparamagnetic Fe3O4@SiO2 nanoparticles as illustrated in Scheme 1.
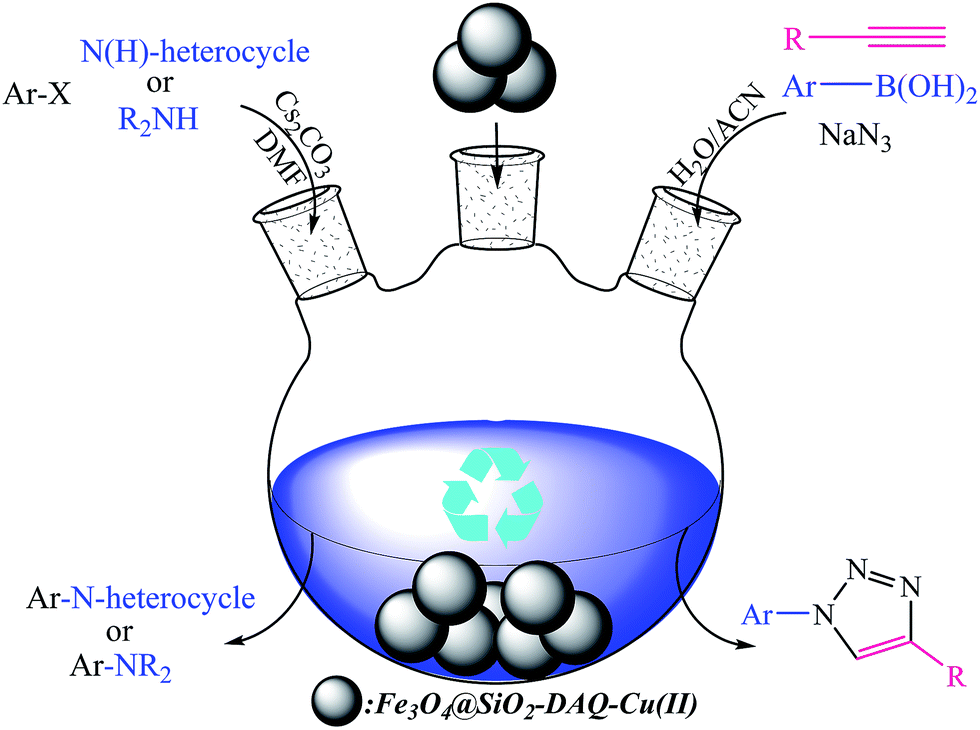 | ||
| Scheme 1 N-Arylation of nitrogen heterocycles and alkylamines with aryl halides and click synthesis of 1-aryl-1,2,3-triazole derivatives is using the Fe3O4@SiO2–DAQ–Cu(II) nanoparticles. | ||
2. Results and discussion
First of all, the preparation of magnetic nanocatalyst with core–shell structure was presented by using nano Fe3O4 as the core, TEOS as the silica source and PVA as the surfactant. Then, Fe3O4@SiO2 was coated with 1,4-dihydroxyanthraquinone–copper(II) nanoparticles.57 The Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2–DAQ–Cu(II) nanocatalysts were characterized by various methods such as Fourier transform infrared spectroscopy (FT-IR), X-ray diffraction analysis (XRD), transmission electron microscopy (TEM), field emission scanning electron microscopy (FE-SEM), dynamic light scattering (DLS), thermogravimetric analysis (TGA), vibration sample magnetometer (VSM), X-ray photoelectron spectroscopic (XPS), N2 adsorption–desorption isotherm analysis and inductively coupled plasma analyzer (ICP).57To optimize the conditions for the N-arylation reaction, initially the reaction between iodobenzene and imidazole as the model reaction was examined in the presence of various amount of the catalysts and the results are presented in Table 1.
| Entry | Catalyst amount (mol%) | Solvent | Base | Temp (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: iodobenzene (1 mmol), imidazole (1.2 mmol), base (2 mmol), Fe3O4@SiO2–DAQ–Cu(II) catalyst and solvent (3 mL). | ||||||
| 1 | 0.45 mol% | EtOH | Cs2CO3 | Reflux | 8 | 21 |
| 2 | 0.45 mol% | MeOH | Cs2CO3 | Reflux | 8 | 29 |
| 3 | 0.45 mol% | H2O | Cs2CO3 | Reflux | 8 | 0 |
| 4 | 0.45 mol% | MeCN | Cs2CO3 | Reflux | 8 | 56 |
| 5 | 0.45 mol% | DMF | Cs2CO3 | 100 °C | 4 | 92 |
| 6 | 0.45 mol% | DMSO | Cs2CO3 | 100 °C | 4 | 89 |
| 7 | 0.45 mol% | Toluene | Cs2CO3 | 110 °C | 8 | 75 |
| 8 | None | DMF | Cs2CO3 | 100 °C | 8 | 0 |
| 9 | 0.2 mol% | DMF | Cs2CO3 | 100 °C | 8 | 34 |
| 10 | 0.3 mol% | DMF | Cs2CO3 | 100 °C | 8 | 69 |
| 11 | 0.4 mol% | DMF | Cs2CO3 | 100 °C | 4 | 84 |
| 12 | 0.5 mol% | DMF | Cs2CO3 | 100 °C | 4 | 91 |
| 13 | 0.45 mol% | DMF | K2CO3 | 100 °C | 8 | 63 |
| 14 | 0.45 mol% | DMF | K3PO4 | 100 °C | 8 | 71 |
| 15 | 0.45 mol% | DMF | Et3N | 100 °C | 8 | 26 |
| 16 | 0.45 mol% | DMF | NaOAc | 100 °C | 8 | 37 |
| 17 | 0.45 mol% | DMF | NaOH | 100 °C | 8 | 65 |
| 18 | 0.45 mol% | DMF | Cs2CO3 | r.t | 8 | Trace |
| 19 | 0.45 mol% | DMF | Cs2CO3 | 60 °C | 8 | 42 |
| 20 | 0.45 mol% | DMF | Cs2CO3 | 80 °C | 6 | 74 |
| 21 | 0.45 mol% | DMF | Cs2CO3 | 90 °C | 4 | 82 |
| 22 | 0.45 mol% | DMF | Cs2CO3 | 110 °C | 4 | 90 |
The results show clearly that catalyst is effective for this transformation and in the absence of it; the reaction did not take place even after higher reaction time (Table 1, entry 8). The use of a lower catalyst loading resulted in incomplete reaction (Table 1, entries 9–11). Increasing the amount of catalyst gave no substantial improvement in the yield (Table 1, entry 12).
Among all the solvents tested, including EtOH, MeOH, H2O, MeCN, DMF, DMSO and toluene, DMF gave the highest yield (Table 1, entries 1–7). The screening of the reaction time indicates that 4 h reaction time is the most suitable to afford the highest yield of product 3a (Table 1, entry 5). We then decided to screen various bases. In general, we obtained superior results with cesium carbonate (Cs2CO3). Replacement of cesium carbonate by potassium carbonate (K2CO3), tripotassium phosphate (K3PO4), triethylamine (Et3N), sodium acetate (NaOAc) or sodium hydroxide resulted in significantly decreased yields (Table 1, entries 5, 13–17).
Also, during our optimization studies, various temperatures were examined and it was found that the temperature plays a significant role in terms of reaction rate and isolated yield and the best results were obtained at 100 °C (Table 1, entries 5, 18–22). Thus, the optimum conditions were found to be 1.0 mmol of iodobenzene, 1.2 mmol of imidazole, 0.45 mol% of the Fe3O4@SiO2–DAQ–Cu(II) catalyst and 2 mmol of Cs2CO3 in DMF (3 mL) at 100 °C.
To examine the scope for this coupling reaction, we have investigated the reactions using a variety of aryl halides and a wide range of N(H)-heterocycles and amines as the substrates under the optimized reaction conditions and the results are outlined in Table 2. We found that aryliodides and aryl bromides containing electron releasing groups (3b, 3e) as well as electron withdrawing groups (3c, 3g) reacted with imidazole to give corresponding N-arylated imidazoles. Presence of electron-withdrawing group such as nitro groups on aryl halides (Table 2, 3c, 3f) increased the yield of the coupling reaction whereas an electron releasing groups such as methyl and methoxy groups on aryl halides (Table 2, 3b, 3e, 3i, 3m) at para position decreased reaction yield for the N-arylation reaction. To test further the scope of this catalyst, reactions with indole, 2-methyl-1H-indole, 1H-benzimidazole and 1H-pyrrole are conducted, which resulted in a very good yield (Table 2, 3h–3o). When diamines such as piperazine were employed, the reaction was complete within 6 h affording 81% yield (Table 2, 3p). Moreover, aryliodides underwent N-arylation of aliphatic amines such as piperazine, phenylpiperazine, dibutylamine and diethylamine under similar conditions to afford the corresponding products in high yields (Table 2, 3p–3t). Further we have screened the chloro-derivatives for the N-arylation of imidazole (Table 2, 3u–3w). N-Arylation of imidazole with chlorobenzene gave no coupled product under the above conditions. However, an increase of catalyst loading (0.6 mol%) with longer reaction time at 130 °C provided 43% yield of the coupled product (Table 2, 3u). Among chloro-derivatives, p-nitrochlorobenzene with a strong electron-withdrawing group resulted in quantitative yield (Table 2, 3v), whereas 2-chloropyridine resulted in lower yield (Table 2, 3w).
|
|---|
| a Reaction conditions: aryl halide (1 mmol), amine (1.2 mmol), Cs2CO3 (2 mmol), Fe3O4@SiO2–DAQ–Cu(II) catalyst (0.45 mol%), DMF (3 mL), 100 °C.b Reaction conditions: aryl halide (2 mmol), amine (1.2 mmol), Cs2CO3 (3 mmol), Fe3O4@SiO2–DAQ–Cu(II) catalyst (0.8 mol%), DMF (5 mL), 100 °C.c Reaction temperature: 130 °C.d Reaction with 0.6 mol% of Fe3O4@SiO2–DAQ–Cu(II) catalyst.e Isolated yield. |
 |

For optimization of the reaction parameters for efficiently synthesis of 1-aryl-1,2,3-triazole derivatives, the reaction of phenyl acetylene (1.1 mmol), phenylboronic acid (1 mmol) and sodium azide (2 mmol) were investigated in the presence of Fe3O4@SiO2–DAQ–Cu(II) as a catalyst in various solvents and also in the presence of various amounts of catalyst. The results are summarized in Table 3. The optimized reaction conditions were: 1.1 mmol of phenyl acetylene, 1.0 mmol of phenylboronic acids, 2.0 mmol of sodium azide, 0.5 mol% of Fe3O4@SiO2–DAQ–Cu(II) catalyst and 3 mL of H2O/ACN (Table 3, entry 11).
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: phenylboronic acid (1 mmol), sodium azide (2 mmol), phenyl acetylene (1.1 mmol), Fe3O4@SiO2–DAQ–Cu(II) catalyst and solvent (3 mL).b Isolated yield. | ||||
| 1 | 0.5 mol% | CH3CH2OH | 10 | 82 |
| 2 | 0.5 mol% | CH3OH | 10 | 78 |
| 3 | 0.5 mol% | i-PrOH | 10 | 35 |
| 4 | 0.5 mol% | CH2Cl2 | 10 | 36 |
| 5 | 0.5 mol% | CH3CN | 10 | 78 |
| 6 | 0.5 mol% | DMSO | 10 | 71 |
| 7 | 0.5 mol% | DMF | 10 | 67 |
| 8 | 0.5 mol% | THF | 10 | 26 |
| 9 | 0.5 mol% | Toluene | 10 | 33 |
| 10 | 0.5 mol% | H2O | 10 | 82 |
| 11 | 0.5 mol% | H2O/ACN | 4 | 95 |
| 12 | None | H2O/ACN | 24 | — |
| 13 | 0.1 mol% | H2O/ACN | 10 | 31 |
| 14 | 0.2 mol% | H2O/ACN | 10 | 64 |
| 15 | 0.3 mol% | H2O/ACN | 6 | 81 |
| 16 | 0.4 mol% | H2O/ACN | 4 | 91 |
| 17 | 0.6 mol% | H2O/ACN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
5 | 92 |
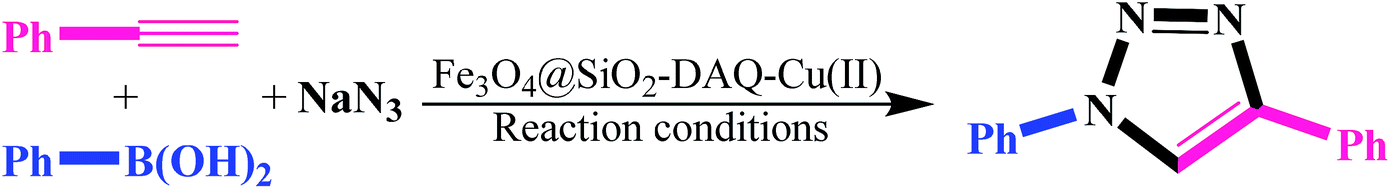
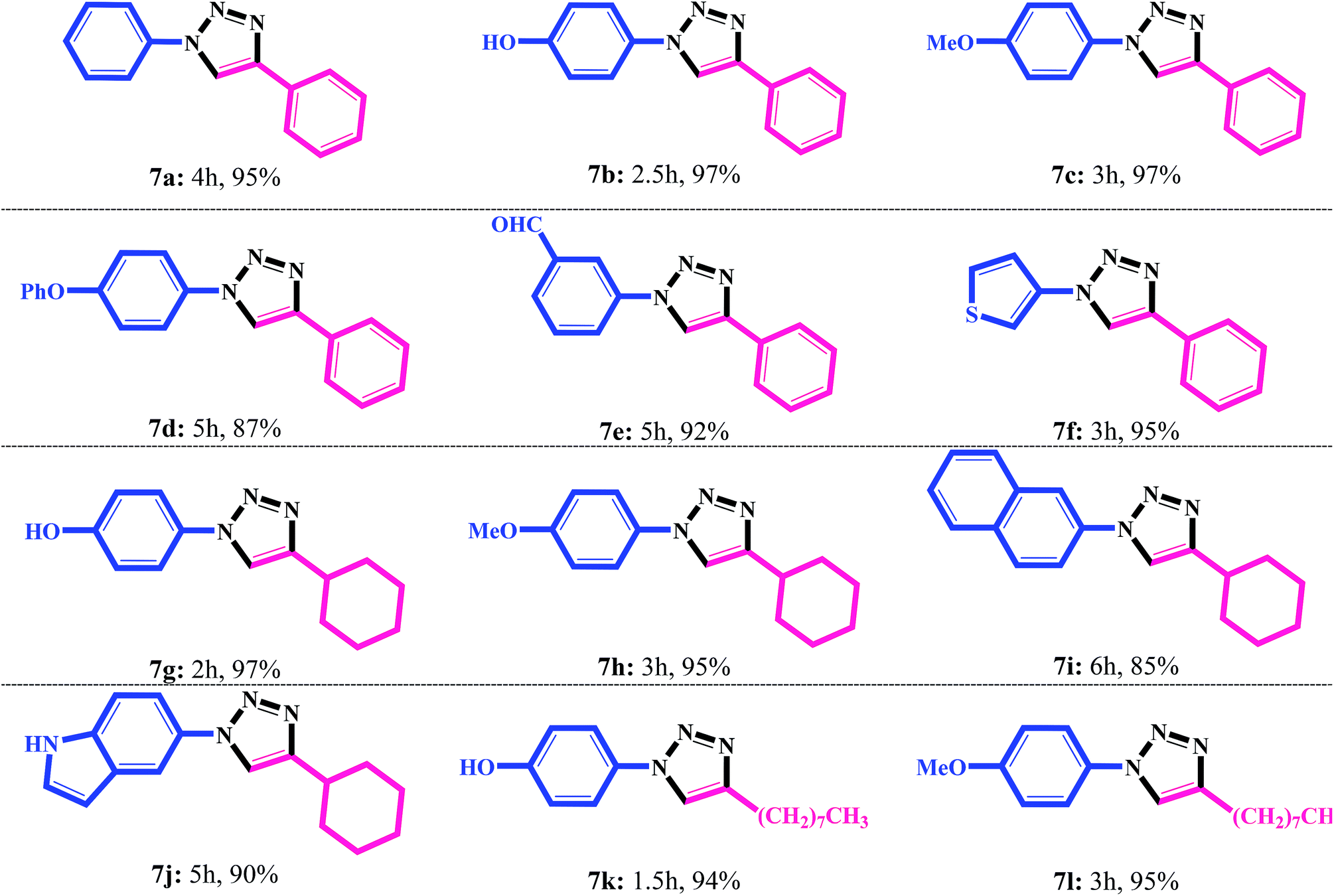
After optimizing the reaction conditions, scope of the Fe3O4@SiO2–DAQ–Cu(II) catalyzed one-pot CuAAC was investigated for variety of boronic acids and alkynes. The results are summarized in Table 4. Various substituted aromatic and heteroaromatic boronic acids and different aromatic and aliphatic alkynes were investigated (Table 4). It was noticed that the triazole products were formed in excellent yields with varying reaction times. The substrate scope of on pot synthesis clearly shows that the reaction time depended on both aryl boronic acid as well as alkyne (Table 4). However, reaction of phenyl boronic acid with phenyl acetylene required 4 h for completion of the reaction (Table 4, 7a). 4-Phenoxyphenyl boronic acids as well as formyl substituted phenyl boronic acid produced corresponding triazoles in 5 h (Table 4, 7d, e). In contrast, as seen in Table 4, the present method is not only suitable for aromatic alkynes but can also successfully be applied to aliphatic alkynes (7g–l). Heteroaromatic boronic acids such as thiophen-3-ylboronic acid and indol-5-yl boronic acid participated well in these reactions producing corresponding triazoles in excellent yields (Table 4, 7f, j).
|
|---|
| a Reaction conditions: aryl boronic acid (1 mmol), sodium azide (2 mmol), alkyne (1.1 mmol), Fe3O4@SiO2–DAQ–Cu(II) catalyst (0.5 mol%), H2O/ACN (1:1), rt. |
 |

A mechanism for the catalytic activity of Fe3O4@SiO2–DAQ–Cu(II) in the synthesis of 1-aryl-1,2,3-triazole derivatives is shown in Scheme 2. The mechanism of CuAAC revealed that sodium azide, which is used as azidonating reagent in one-pot protocol reduces Cu(II) to click-active Cu(I).58 Thus, the plausible mechanism for one-pot Fe3O4@SiO2–DAQ–Cu(II) catalyzed CuAAC reaction is depicted in the Scheme 2.
 | ||
| Scheme 2 Plausible mechanism of one-pot synthesis of 1-aryl-1,2,3-triazoles using Fe3O4@SiO2–DAQ–Cu(II) catalyst. | ||
The recycling of the catalyst is an important issue in heterogeneous catalysis system. Thus, we turn our attention to reusability of our Fe3O4@SiO2–DAQ–Cu(II) catalyst in the preparation of 1-phenyl-1H-imidazole (3a) and 1,4-diphenyl-1H-1,2,3-triazole (7a) under optimized conditions. It is noteworthy that the catalyst could be used six times without significance loss of its activity (Fig. 1a). Thus, it is reasonable to believe that the immobilized catalyst can be repeatedly used for large-scale production without significant loss of its catalytic activity.
 | ||
| Fig. 1 (a) Recyclability of Fe3O4@SiO2–DAQ–Cu(II) in the synthesis of 3a and 7a under the optimized conditions; (b) photograph of a magnet attracting Fe3O4@SiO2–DAQ–Cu(II) in solution; (c) and (d) TEM and DLS images of Fe3O4@SiO2–DAQ–Cu(II) nanoparticles after six reaction cycles. | ||
The superparamagnetic property of the particle is very important for its application.
As shown in Fig. 1b, the magnetic Fe3O4@SiO2–DAQ–Cu(II) particles in solution were black emulsion, and it could be separated from solution in a short period under an external magnetic field. Thus, the attraction and dispersion processes can be readily altered by applying and removing an external magnetic field, showing good dispersion and magnetic separation.
Also, the morphology of the recycled Fe3O4@SiO2–DAQ–Cu(II) particles after six runs was analyzed. Fig. 1c show representative TEM image of the catalyst after 6 cycles of catalytic reaction. As revealed by TEM, the obtained magnetite particles possess approximately spherical shapes and a mean diameter of 60 nm (Fig. 1c).
We did not observe significant change in the morphology of the catalyst, but some particles might have aggregated. DLS measurements were also carried out to measure the hydrodynamic diameter of the nanoparticles after six reaction cycles. This size distribution is centered at a value of 70 nm (Fig. 1d). Generally, the size of nanoparticles will be increased after each cycle and leaching of copper, and increasing of catalyst size lead to a decrease in the yield.
To determine the exact species responsible for the observed reactions and to measure the extent of Cu leaching after the reactions, we have used the hot filtration test.59,60 For this aim, we have studied the reaction of iodobenzene, imidazole and cesium carbonate in the presence of Fe3O4@SiO2–DAQ–Cu(II) catalyst under optimized conditions. When the reaction was half complete, the nanoparticles were removed in situ at the reaction temperature, and the reactants were allowed to undergo further reaction in the solution. We confirmed that there was no further conversion of the reactants in the absence of the nanoparticles. These results indicate that there was little of the active metal species in the solution phase and confirmed that the catalyst acts heterogeneously in the reaction.
Also, in order to determine the absolute amount of the copper species dissolved in solution caused by leaching, the crude reaction mixtures were evaporated to dryness and analyzed using Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES). It was shown that 2.67 wt% (0.0061 mmol g−1) of the total amount of the original copper species was lost into solution during the course of the reaction after six cycles. Therefore, the analysis of the reaction mixture by the ICP technique showed that the leaching of Cu was negligible.
3. Conclusion
In conclusion, we developed a mild and practical Fe3O4@SiO2–DAQ–Cu(II) catalyzed N-arylation of N(H)-heterocycles and alkylamines with aryl halides to afford corresponding coupled products in good to excellent yields without using any external ligands or additives as promoters. N-Arylated products were isolated in good to excellent yields, demonstrating the versatility of the protocol. This heterogeneous catalyst, also efficiently works for the efficient triazole synthesis stating from NaN3, alkynes, and aryl boronic acid derivatives in H2O/ACN at room temperature. Heterogeneous nature, thermal stability, easy separation of the catalyst, simple procedure, excellent yields, short reaction time, easy product separation and lower loading of catalyst makes this method an instrumental alternative to the previous methodologies for the scale up of these reactions. In addition, these methods offer the competitiveness of recyclability of the catalyst without significant loss of catalytic activity, and the catalyst could be easily recovered by external magnetic field and reused for at least 6 cycles.4. Experimental
4.1. Materials and physical measurements
All chemicals were of analytical grade and used as received. The progress of the reactions was followed by TLC using silica gel SILG/UV 254 plates. IR spectra were recorded with KBr pellets using a Shimadzu 8300 spectrophotometer. X-ray diffraction (XRD) pattern was recorded on a Bruker AXS D8-advance X-ray diffractometer using Cu Kα radiation (λ = 1.5418 Å). The morphology and size of the particles were examined by transmission electron microscopy (TEM) using a Philips EM208 transmission electron microscope. Field emission scanning electron microscopy (FE-SEM) was recorded on a Hitachi S-4160 instrument.Particle sizes of MNPs were measured using a HORIBA-LB550 dynamic light scattering. The X-ray photoelectron spectra (XPS) were recorded on the XR3E2 (VG Microtech) twin anode X-ray source using AlKα = 1486.6 eV. The 1H and 13C NMR spectra were recorded on a Bruker Avance DPX 250 MHz spectrometer (using CDCl3 with TMS as the standard). The Cu analysis and leaching test was carried out by ICP analyzer (Varian, Vista-pro). The element analyses (C, H, N) were obtained from a Thermo finigan Flash EA-1112 CHNSO rapid elemental analyzer. Melting points were recorded with an Electrothermal Type 9100 melting point apparatus. Therefore, all of the products were characterized by FT-IR, 1H NMR and 13C NMR, and also by comparison with authentic samples.
4.2. General procedure
Then, Fe3O4@SiO2 core–shell nanospheres were prepared through a modified Stöber method.62 In a typical process, 0.05 g of synthesized Fe3O4 nanoparticles were dispersed in solution composed of 5 mL of deionized water, 50 mL of ethanol and 5 mL of NaOH (10 wt%). Subsequently, 0.2 mL TEOS (tetraethoxysilane) was slowly added and the mixture was stirred vigorously. After being stirring for 30 min, the Fe3O4@SiO2 nanoparticles were magnetically collected, washed with ethanol and deionized water, and then dried under vacuum at 80 °C for 10 h.
:nhexane (1:1) to afford pure compound.60:1) (3.0 mL) were added and the mixture was stirred at room temperature. The progress of the reaction was monitored by thin layer chromatography (TLC). After stirring for the appropriate time, the catalyst was removed using magnetic field. Then, the filtrate was extracted with EtOAc (3 × 10 mL) and dried over anhydrous sodium sulfate. Combined organic layer was evaporated to dryness, and the solid residue recrystallized from EtOH/H2O (1:3 v/v) to give a crystalline pure product.4.3. Spectral data
Acknowledgements
This work was supported by council of Iran National Science Foundation and University of Shiraz from the Iran Society for the Promotion of Science.References
- S. U. Son, Y. J. Jang, J. Park, H. B. Na, H. M. Park, H. J. Yun, J. Lee and T. Hyeon, J. Am. Chem. Soc., 2004, 126, 5026 CrossRef CAS PubMed.
- H. M. Chen, C. K. Chen, R. S. Liu, L. Zhang, J. J. Zhang and D. P. Wilkinson, Chem. Soc. Rev., 2012, 41, 5654 RSC.
- Z. J. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321 CrossRef CAS PubMed.
- M. Esmaeilpour, A. R. Sardarian and J. Javidi, Catal. Sci. Technol., 2016, 6, 4005 CAS.
- P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435 RSC.
- Y. Zhu, L. P. Stubbs, F. Ho, R. Liu, C. P. Ship, J. A. Maguire and N. S. Hosmane, ChemCatChem, 2010, 2, 365 CrossRef CAS.
- M. Esmaeilpour, J. Javidi, F. Dehghani and F. Nowroozi Dodeji, New J. Chem., 2014, 38, 5453 RSC.
- S. Ko and J. Jang, Angew. Chem., 2006, 118, 7726 CrossRef.
- P. Li, L. Wang, L. Zhang and G. W. Wang, Adv. Synth. Catal., 2012, 354, 1307 CrossRef CAS.
- M. Esmaeilpour, J. Javidi and M. Zandi, Mater. Res. Bull., 2014, 55, 78 CrossRef CAS.
- M. Cai, J. Peng, W. Hao and G. Ding, Green Chem., 2011, 13, 190 RSC.
- Y. M. A. Yamada, K. Takeda, H. Takahashi and S. Ikegami, J. Org. Chem., 2003, 68, 7733 CrossRef CAS PubMed.
- J. Liu, X. Huo, T. Li, Z. Yang, P. Xi, Z. Wang and B. Wang, Chem.–Eur. J., 2014, 20, 11549 CrossRef CAS PubMed.
- N. Audic, H. Clavier, M. Mauduit and J. C. Guillemin, J. Am. Chem. Soc., 2003, 125, 9248 CrossRef CAS PubMed.
- A. Schatz, M. Hager and O. Reiser, Adv. Funct. Mater., 2009, 19, 2109 CrossRef.
- M. Esmaeilpour, J. Javidi, F. Dehghani and F. Nowroozi Dodeji, RSC Adv., 2015, 5, 26625 RSC.
- D. Zhang, C. Zhou, Z. Sun, L. Z. Wu, C. H. Tung and T. Zhang, Nanoscale, 2012, 4, 6244 RSC.
- M. B. Gawande, P. S. Brancoa and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371 RSC.
- M. Esmaeilpour, J. Javidi and M. Zandi, New J. Chem., 2015, 39, 3388 RSC.
- M. M. J. Modo and J. W. M. Bulte, Molecular and cellular MR imaging, CRC Press, Boca Raton, 2007 Search PubMed.
- S. Boutry, S. Laurent, L. V. Elst and R. N. Muller, Contrast Media Mol. Imaging, 2006, 1, 15 CrossRef CAS PubMed.
- J. Javidi and M. Esmaeilpour, Colloids Surf., B, 2013, 102, 265 CrossRef PubMed.
- M. M. Miller, G. A. Prinz, S. F. Cheng and S. Bounnak, Appl. Phys. Lett., 2002, 81, 2211 CrossRef CAS.
- H. Pardoe, P. R. Clark, T. G. St Pierre, P. Moroz and S. K. A. Jones, Magn. Reson. Imaging, 2003, 21, 483 CrossRef CAS PubMed.
- M. Esmaeilpour, A. R. Sardarian, A. Jarrahpour, E. Ebrahimi and J. Javidi, RSC Adv., 2016, 6, 43376 RSC.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef CAS PubMed.
- J. Javidi, M. Esmaeilpour and M. Rajabnia Khansari, RSC Adv., 2015, 5, 73268 RSC.
- M. Esmaeilpour, J. Javidi, F. Nowroozi Dodeji and M. Mokhtari Abarghoui, Transition Met. Chem., 2014, 39, 797 CrossRef CAS.
- M. Esmaeilpour and J. Javidi, J. Chin. Chem. Soc., 2015, 62, 614 CrossRef CAS.
- D. Kim, N. Lee, M. Park, B. Y. Kim, K. An and T. Hyeon, J. Am. Chem. Soc., 2009, 131, 454 CrossRef CAS PubMed.
- R. Xiong, Y. Wang, X. Zhang, C. Lu and L. Lan, RSC Adv., 2014, 4, 6454 RSC.
- Z. Markova, K. Siskova, J. Filip, K. Safarova, R. Prucek, A. Panacek, M. Kolar and R. Zboril, Green Chem., 2012, 14, 2550 RSC.
- M. L. Quan, P. Y. S. Lam, Q. Han, D. J. P. Pinto, M. Y. He, R. Li, C. D. Ellis, C. G. Clark, C. A. Teleha, J. H. Sun, R. S. Alexander, S. Bai, J. M. Luettgen, R. M. Knabb, P. C. Wong and P. R. Wexler, J. Med. Chem., 2005, 48, 1729 CrossRef CAS PubMed.
- H. Zhang, Q. Cai and D. Ma, J. Org. Chem., 2005, 70, 5164 CrossRef CAS PubMed.
- F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382 CrossRef.
- J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed.
- H. J. Cristau, P. P. Cellier, J. F. Spindler and M. Taillefer, Chem.–Eur. J., 2004, 10, 5607 CrossRef CAS PubMed.
- A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef CAS PubMed.
- H. J. Cristau, P. P. Cellier, J. F. Spindler and M. Taillefer, Eur. J. Org. Chem., 2004, 695 CrossRef CAS.
- D. M. T. Chan, K. L. Monaco, R. Li, D. Bonne, C. G. Clark and P. Y. S. Lam, Tetrahedron Lett., 2003, 44, 3863 CrossRef CAS.
- D. Schweinfurth, S. Strobel and B. Sarkar, Inorg. Chim. Acta, 2011, 374, 253 CrossRef CAS.
- D. Amantini, F. Fringuelli, O. Piermatti, F. Pizzo, E. Zunino and L. Vaccaro, J. Org. Chem., 2005, 70, 6526 CrossRef CAS PubMed.
- Y. Xia, Z. Fan, J. Yao, Q. Liao, W. Li, F. Qu and L. Peng, Bioorg. Med. Chem. Lett., 2006, 16, 2693 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- E. Frank, J. Molnár, I. Zupkó, Z. Kádár and J. Wölfling, Steroids, 2011, 76, 1141–1148 CrossRef CAS PubMed.
- S. Chassaing, A. S. S. Sido, A. Alix, M. Kumarraja, P. Pale and J. Sommer, Chem.–Eur. J., 2008, 14, 6713 CrossRef CAS PubMed.
- B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235 CrossRef CAS PubMed.
- T. Miao and L. Wang, Synthesis, 2008, 363 CAS.
- C. Girard, E. Önen, M. Aufort, S. Beauvière, E. Samson and J. Herscovici, Org. Lett., 2006, 8, 1689 CrossRef CAS PubMed.
- M. Meldal and C. W. Tomøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- J. Y. Kim, J. C. Park, H. Kang, H. Song and K. H. Park, Chem. Commun., 2010, 46, 439 RSC.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS PubMed.
- J. Jlalia, F. Gallier, N. Brodie-Linder, J. Uziel, J. Augé and N. Lubin-Germain, J. Mol. Catal. A: Chem., 2014, 393, 56 CrossRef.
- Y. Masuyama, K. Yoshikawa, N. Suzuki, K. Hara and A. Fukuoka, Tetrahedron Lett., 2011, 52, 6916 CrossRef CAS.
- K. Namitharan, M. Kumarraja and K. Pitchumani, Chem.–Eur. J., 2009, 15, 2755 CrossRef CAS PubMed.
- M. Esmaeilpour and J. Javidi, J. Chin. Chem. Soc., 2015, 62, 328 CrossRef CAS.
- M. Esmaeilpour, J. Javidi and S. Zahmatkesh, Appl. Organomet. Chem., 2016 DOI:10.1002/aoc.3518.
- S. Mohammed, A. K. Padala, B. A. Dar, B. Singh, B. Sreedhar, R. A. Vishwakarma and S. B. Bharate, Tetrahedron, 2012, 68, 8156 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed.
- A. Biffis, M. Zecca and M. Basato, Eur. J. Inorg. Chem., 2001, 1131 CrossRef CAS.
- J. Javidi and M. Esmaeilpour, Mater. Res. Bull., 2016, 73, 409 CrossRef CAS.
- M. Esmaeilpour, J. Javidi, F. Nowroozi Dodeji and H. Hassannezhad, J. Iran. Chem. Soc., 2014, 11, 1703 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16646f |
| This journal is © The Royal Society of Chemistry 2016 |