
A GMDH-type neural network with multi-filter feature selection for the prediction of transition temperatures of bent-core liquid crystals
Davor Antanasijevića,
Jelena Antanasijević*b,
Viktor Pocajtb and
Gordana Ušćumlićb
aInnovation Center of the Faculty of Technology and Metallurgy, Karnegijeva 4, 11120 Belgrade, Serbia
bUniversity of Belgrade, Faculty of Technology and Metallurgy, Karnegijeva 4, 11120 Belgrade, Serbia. E-mail: jantanasijevic@tmf.bg.ac.rs
First published on 13th October 2016
Abstract
A novel strategy for the prediction of the transition temperature of bent-core liquid crystals (LCs) based on the combination of multi filter feature selection and group method of data handling (GMDH) type neural networks is reported. An entire set of 243 compounds was randomly divided into a training set of 207 compounds and a test set of 36 compounds. Descriptors were selected from a pool of 2D, and two pools of 2D and 3D ones, optimized by molecular mechanics (MM) and semi-empirical (SE) method. The reduction of the pool of descriptors was performed using multi filters based on chi square and v-WSH algorithm, while the final subset selection was performed by GMDH algorithm during the learning process. The obtained 2D, MM and SE GMDH models have 11, 13 and 16 descriptors, respectively, and demonstrate good generalization and predictive ability (R2 = 0.92). The final models were subjected to a randomization test for validation purpose. Those models appear to be not only suitable for prediction, but they also allow the identification of key structural features that alter the transition temperature of bent-core LCs.
Introduction
Liquid crystal (LC) molecules share important properties of both liquids and crystals: they flow like a liquid and at the same time maintain some degree of positional and/or orientational order.1 As such, they have unique physicochemical properties and consequently wide application in various fields.2,3 But, in order to be used in any particular technological application, thermotropic LCs have to possess stable mesophases in a suitable temperature range.4 The upper temperature limit (i.e. transition temperature) at which mesophase exists can be used as a measure of its stability.5Quantitative structure–property relationship (QSPR) methodology has been often used to predict various physical and chemical properties of LCs.4–10 Artificial neural networks (ANNs), as a nonlinear modelling approach, are mostly used for this purpose, due to complex relationships exist between a property of molecule and its structure.6 Among first, Johnson and Jurs4 have shown that the clearing temperatures of a series of structurally similar rod-like LCs can be successfully predicted using ANNs. In a recent study, Antanasijević et al. have used QSPR method in combination with ANNs, decision trees (DTs) and MARS (multivariate adaptive regression splines) technique for the prediction of liquid crystallinity,10 and with DT and MARS for the estimation of the clearing temperatures7 of five-ring bent-core molecules.
Feature selection is an important step in QSPR development, concerning that a large number of molecular descriptors (up to two thousands) can be calculated for each structure.11 In general, a large pool of descriptors can be reduced using filter, wrapper or embedded feature selection methods. Filter techniques eliminate irrelevant and redundant features by checking data consistency,12 while wrappers evaluate the usefulness of an input set during the model training.13 Embedded methods perform variable selection in the process of training and they are specific to given learning machines.14 Since filters work much faster, they are suitable for large datasets, while wrapper and embedded methods achieve excellent accuracy at the cost of significant time.15 In recent years, hybrid approaches are proposed in order to combine the advantages of both methods.16,17
It is generally more convenient to have a linear or polynomial QSPR model that enables analysis of particular descriptor contribution and therefore group method of data handling (GMDH) type neural networks can be used as an alternative to standard ANNs, which operates like a ‘black box’ model.18
GMDH is a specific type of feed-forward ANNs, which algorithm was firstly introduced by Ivakhnenko19 and enhanced by others.20 The GMDH-type ANNs, often referred to as polynomial neural networks, are based on the identification of the functional structure of a model, which is extracted from the empirical data by polynomial functions.21 Therefore, a nonphysical model, with high accuracy and simpler structure than a corresponding physical model, can be obtained by applying GMDH on complex (non-linear) input–output relationships, especially if the available dataset is small and noisy inputs are present.22
In the last decade, GMDH has been used to solve complex engineering problems and to identify the behaviour of nonlinear systems in many fields, such as control engineering, data mining, process optimization, and medical image recognition and diagnosis (see studies23–26 and reference cited therein).
In this study, we report the development of QSPR model using GMDH-type neural network for the prediction of the transition temperatures of five-ring bent-core LCs. Although GMDH operates as an embedded feature selection method, chi square ranking and correlation filter were applied in the pre-processing step in order to reduce the pool of descriptors and to enhance the descriptor selection process. To date, this is the first application of GMDH-type neural networks for the prediction of LC properties.
Computational methods
Dataset
A recently published dataset (see Table S1 in the supplemental material of paper by Antanasijević et al.7), which contains transition temperature values for 243 bent-core LC compounds, was utilized for the development and testing of GMDH models. In this dataset the transition temperatures were in the range from 352.15 to 458.15 K. The dataset consisted of structurally diverse five-ring aromatic compounds in the terms of the type of linkage groups and their orientation, substituents on the rings, and the type and length of terminal chains. The same subset of 36 compounds was used for model testing, in order to allow direct comparison with the models created in the previous study.7Structure optimization and descriptor generation
The molecular structures were firstly sketched in ChemDraw software, and then initially optimized using MMFF94 optimization routine (ChemAxon, Marvin27). The final geometry of the minimum energy conformation was obtained using the semi-empirical PM3 method (Polak–Ribiere algorithm) using HyperChem8.0 program.28 The structures were optimized at the restricted Hartree–Fock level until the RMS gradient was 0.01 kcal Å−1 mol−1.In order to check the accuracy of the applied optimisation methods, the obtained structures were compared with available optimized structures from DFT studies. For example, the DFT study29 for the compound 161 indicates that bending angle (α), which determines molecular packing and therefore its transition temperature, has the value of 121°. The α of 125° and 126° obtained in this study for the same compound by MM and SE method, respectively, is in the fair agreement with the abovementioned DFT value.
Subsequently, the calculation of molecular descriptors was performed using PaDEL-Descriptor software.30 After the elimination of descriptors with constant and near constant values, the pool of 501 constitutional, topological, geometric, electrostatic and hybrid descriptors (360 2D and 141 3D) was remained.
Descriptor selection
The feature selection was performed as presented in Fig. 1a: | ||
| Fig. 1 (a) Strategies for the selection of descriptors (b) the number of descriptors selected in each step. CF90 and CF99 stand for correlation filter with r cut-off equal to 0.90 and 0.99, respectively. | ||
(a) The A models were created using correlation filter (CF) in order to eliminate collinear descriptors (r > 0.90), after which GMHD is used to select the best subset of descriptors during learning (embedded feature selection);
(b) The B models were created using multi filter approach that combines a chi square (CS) ranking in the first step with a collinear based elimination of descriptors in the second step, after which, in the third step, GMHD was used as embedded method. Prior the use of CS, near constant and highly correlated (r > 0.99) descriptors were removed in order to reduce redundant and non-useful information.31
The V-WSP variable reduction algorithm, proposed by Ballabio et al.,32 was used as correlation filter. This filter is an adaptation of the WSP (Wootton, Sergent, Phan-Tan-Luu's) algorithm, which was developed for space-filling designs of experiments and has been modified with the aim to select a representative set of variables instead of points.32 A Java implementation of this algorithm (the V-WSP tool) by Ambure et al.33 has been used in this study.
The CS is a supervised univariate feature selection method that ranks the molecular descriptors according to their statistical association with the modelled output, where larger CS values imply more significant descriptors. A CS feature selection implementation in Statistica34 was used, and because the CS is an association measure for categorical variables, the software's default number of bins (ten) was used for the chi square discretizing of molecular descriptors.35
The final step in the both feature selection approaches is the application of GMDH, which has been proved to be effective with neural network classifiers.12
GMDH-type neural network
The GMDH algorithm, details of which can be found in literature,36 differs from standard regression analysis and it is “similar to the way in which nature evolves by natural selection”.37 There is a variety of supervised GMDH algorithms:38 combinatorial algorithm, multilayered iterative algorithm (MIA), harmonical algorithm, objective system analysis, etc. Also, several enhancements related to the determination of structure, parameters and uncertainty of the GMDH models have been proposed in the recent years,39,40 in order to increase their effectiveness for certain tasks. For example, unscented Kalman filter approach was applied for the design of GMDH model and determination of its uncertainty in order to obtain robust sensor and actuator for fault detection and diagnosis.41,42In this study, the MIA variant of GMDH that is implemented in NeuroShell 2 (ref. 43) was used. This is a self-organizing algorithm that uses the best polynomial terms (so-called “survivors”) from the first layer (eqn (1) and (2)) obtained by regressing pair of inputs (e.g. x1 and x2), as arguments in the next layer (eqn (3)).
The first layer
| y1 = a10 + a11x1 + a12x2 + a13x1x2 | (1) |
| y2 = a20 + a21x3 + a22x4 + a23x3x4 | (2) |
The next layer
| z1 = b0 + b1y1 + b2y2 + b3y1y2 | (3) |
As can be observed, the original inputs can be propagated thought the network without a construction of their polynomial form, which can reduce overall model complexity.
The layers were built until a certain stopping criterion was met. Over-fitting can be prevented using cross-validation or a statistical metric that penalize model complexity. In this study, the prediction squared error (PSE), introduced by Barron,44 was applied as a stopping criterion, see eqn (4), where To is the observed temperature, Tp is predicted temperature, σo is output variance, k is the number of model parameters and Np is the number of training data points.
 | (4) |
In comparison with standard neural networks, the GMDH architecture (Fig. 2) is being fully adjusted both structurally and parametrically during training.45 It is composed of an input layer, several hidden layers and an output layer. The number of input neurons is equal to the number of inputs, while each hidden layer consists of one or more neurons. Each hidden neuron is actually the resultant network that processes two inputs and generate one polynomial term.
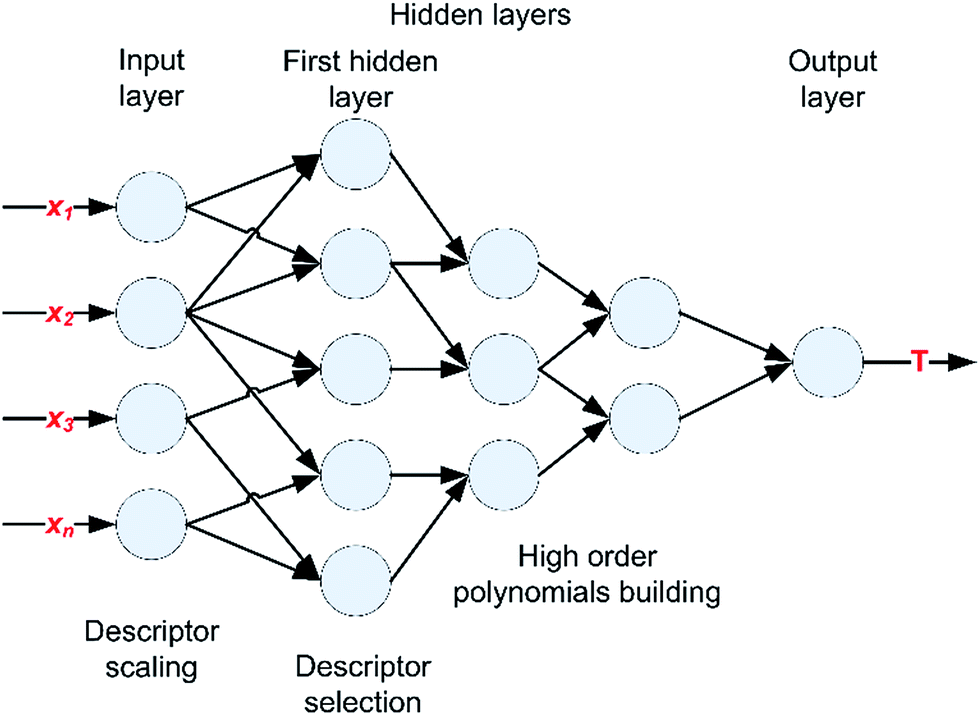 | ||
| Fig. 2 A typical architecture of GMDH-type neural network. | ||
The input layer scales the descriptor values, while the first hidden layer performs the selection of descriptors. In the second, third and etc. hidden layers, higher order polynomials are being built. Since the number of survivors in the first hidden layer affects the diversity of final polynomial model and the quality of choice of important variables, the optimal maximum number of survivors (NmaxL1S) that are propagated to the second hidden layer needs to be defined.
NmaxL1S depends on the complexity of the problem, as well on the number of inputs presented to the GMDH. In order to empirically determine the optimal value of NmaxL1S in respect to the number of inputs, initial simulations with 2D descriptors were performed. The software that was used limits the value of NmaxL1S to 100, while in the case where the number of inputs (Ni) is lower than 50, the NmaxL1S is limited to twice Ni. The results obtained on cross-testing (with two datasets where each contained 20% of the original dataset) and NmaxL1S dependence of Ni are presented in Fig. 3. As can be observed, the dependence of NmaxL1S on Ni decreases with the increase of Ni, and can be approximated with linear/constant relationships in four regions.
 | ||
| Fig. 3 (a) Cross testing results (RCT2) with datasets containing different numbers of inputs (b) the maximum number of polynomials (NmaxL1S) in the first layer of GMDH network depending on the number of inputs (Ni). | ||
Other GMDH parameters that need to be defined prior to the training are the maximum number of descriptors in polynomials term, which was set to 4 for linear and to 3 for all other terms, and degree of polynomials, which was set to 3.
Results and discussion
The comparison of models
Both feature selection approaches were performed separately for each pool of descriptors (Fig. 1b). The number of CS ranked descriptors (150 and 200) that were to be further used was set to be about 50% higher that the number of descriptors that have remained after the application of correlation filter in the case of A models. The lowest ranked descriptor selected by GMDH in the case of 2D model had the rank of 136, while in the case of MM and SE the rank was 195 and 168, respectively. All three B models used lower number of descriptors in comparison with the corresponding A models.The obtained A and B models were evaluated using Taylor diagram (Fig. 4). Taylor diagrams46 provide a concise statistical summary of how well different models perform in the terms of their correlation (r), centered root mean square error (RMSE), and amplitude of their variations (standard deviations). Those three metrics are plotted simultaneously in the two-dimensional space using the following equation:
| E2 = σo2 + σp2 − 2σo2σp2r | (5) |
 | (6) |
 | ||
| Fig. 4 Taylor diagram for the A and B GMDH models. | ||
In Fig. 4, it can be easily observed that the B models have lower error (i.e. centered RMSE) and higher correlation in comparison with the A models. Therefore, the applied multi filtered feature selection provides more accurate GMDH models than those obtained using the single correlation filter.
Regarding the B models, it can be seen that the models 2D and MM have almost the same centered RMSE and correlation, while the SE model has the standard deviation very similar to the observed one. In the next section, a detailed evaluation of the performance of B models is presented.
The GMDH parameters and performance metrics for B models are summarized in Table 1. The pool of 2D descriptors has been reduced to 19% of its initial size (from 360 to 70) after the multi filter feature selection was applied. A similar reduction of approximately 80% was obtained for the 2 & 3D models as well. All three models have the same number of hidden layers, but use a different number of descriptors which seems to correspond with the size of initial pool. The adjusted R2 (0.92) and RMSE (6.68 ± 0.14 K) demonstrate that the generalization of all three QSPR models is statistically stable and that the models fit the test data well. The MM model performed slightly better, with the RMSE of 6.52 K, which is an improvement of 0.9 K in comparison with the results obtained in previous study7 using the MARS technique.
| Pool of descriptors | Type | 2D | MM (2 & 3D) | SE (2 & 3D) | |
|---|---|---|---|---|---|
| a Number of hidden layers.b To access descriptors see Table 2. | |||||
| Ni | 70 | 104 | 101 | ||
| GMDH parameters | NmaxL1S | 62 | 66 | 66 | |
| NLa | 8 | 8 | 8 | ||
| Descriptors usedb (pool reduction) | 11 (84%) | 13 (88%) | 16 (84%) | ||
| Performance metrics | Adjusted R2 | 0.920 | 0.922 | 0.916 | |
| F | 401.7 | 414.7 | 372.9 | ||
| RMSE (K) | 6.66 | 6.52 | 6.87 | ||
| Absolute error (K) | Min. | 0.021 | 0.028 | 0.006 | |
| Mean | 5.15 | 4.89 | 5.02 | ||
| Max. | 19.4 | 15.6 | 18.5 | ||
The Y-randomization was performed as an additional validation step in order to obtain an estimate of chance correlation.47–52 The measured transition temperatures were shuffled by 10 random exchanges in their positions for each model, while the descriptors matrix has remained unchanged. In order to include the “selection bias”, as suggested by Rücker et al.,53 randomized GMDH models were created using the same pool of descriptors and network parameters as the real ones. The risk of chance correlation was quantified by the value of Rp2 that is calculated from the eqn (7) in which Rr2 describes the training performance of randomized models, while R2 stands for real QSPR models.54 For a QSPR model having Rp2 > 0.5, it may be considered that the model has not been obtained by chance alone.55
 | (7) |
For all models, the Rp2 value was higher than 0.5 (Fig. 5), which indicates that they have passed the randomization test.
 | ||
| Fig. 5 Y-Randomization results. | ||
Also, the real GMDH models have R2 higher than the corresponding randomized models by more than 3 standard deviations (SD) (16 SD for 2D, 8 SD for MM and 11 SD for SE), which confirms their statistical significance at the 0.1% level.53
As expected, the randomization results have shown that the risk of randomly obtained correlation increases with the size of pool of descriptors. Therefore, GMDH should be applied to the lowest possible pool of descriptors, and results suggest that the critical value is 115 descriptors (Fig. 5). On the other hand, it should be noted that the randomly correlated GMDH models can be easily identified, since all of them have more than 30 hidden layers and very complex polynomial equations.
The correlation between the experimental and predicted transition temperatures is shown graphically in Fig. 6a, where the outliers are also labelled. As can be seen from Fig. 6a, the compound 40 is an outlier in all three models, while the compound 42, which is from the same series, is an outlier only in the case of 2D model, but its transition temperature was also predicted with a higher error by both 2 & 3D models. Compounds 40 and 42 are from the series 38–42, which is structurally very similar to the series 29–37, the only difference being the orientation of the azomethine group (Fig. 6b). This small structural variation significantly alters the transition temperatures, i.e. corresponding homologues differ up to 30 K. This effect has not been captured by the selected descriptors, thus the predicted transition temperatures of those compounds correspond to their homologues from the series 29–37, which was more prevalent in the training set.
 | ||
| Fig. 6 (a) Measured vs. predicted plots with outliers. Solid lines represent the line of slope 1, while dashed lines indicate 3 SD error, (b) structure of outliers from the series 38–42, (c) structure and transition temperature of compounds from the series 50–58. | ||
Regarding the compound 50, it exhibits an unexpectedly high transition temperature in comparison with its homologues from the same series (Fig. 6c), while in the case of compound 87 no obvious reason can be found for it to be an outlier.
The interpretation of descriptors
The eqn (8)–(10) were obtained using GMDH method with the pool of descriptors reduced by multi filter feature selection approach. In those equations, the descriptors are labelled according to the group they belong (Table 2), while in addition the 3D ones are marked with bold letters.
 | (8) |
 | (9) |
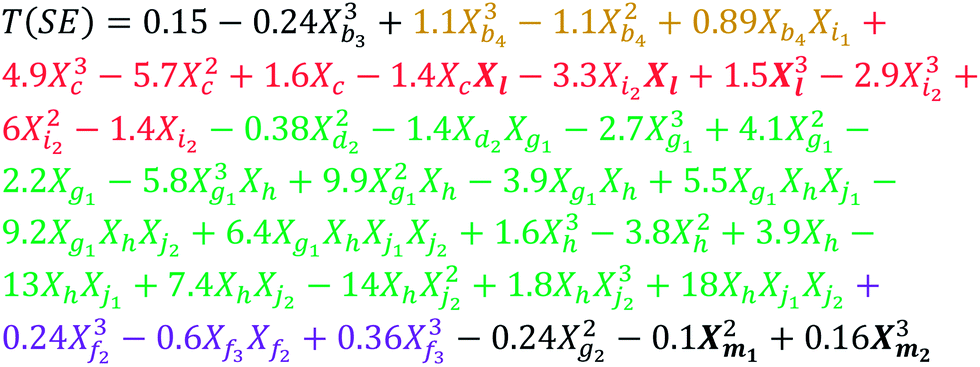 | (10) |
| Group | Label (Eq. symbol) | Description |
|---|---|---|
| a Burden – CAS – University of Texas eigenvalue.b Molecular linear free energy relation.c Charged partial surface area.d Weighted holistic invariant molecular. | ||
| ALOGP | AlogP (Xa) | Ghose-Crippen LogKow |
| Barysz matrix | SM1_Dzi (Xb1) | Spectral moment of order 1 weighted by first ionization potential |
| SM1_DzZ (Xb2) | Spectral moment of order 1 weighted by atomic number | |
| VR2_Dzs (Xb3) | Normalized Randic-like eigenvector-based index weighted by I-state | |
| VE1_Dzp (Xb4) | Coefficient sum of the last eigenvector weighted by polarizabilities | |
| BCUTa | BCUTp-1l (Xc) | High lowest polarizability weighted BCUTS |
| Carbon types | C3SP2 (Xd1) | Doubly bound carbon bound to three other carbons |
| C2SP2 (Xd2) | Doubly bound carbon bound to two other carbons | |
| Chi path cluster | VPC-4 (Xe) | Valence path cluster, order 4 |
| Information content | TIC5 (Xf1) | Total information content index (neighborhood symmetry of 5-order) |
| CIC1 (Xf2) | Complementary information content index (neighborhood sym. of 1-order) | |
| MIC0 (Xf3) | Modified information content index (neighborhood symmetry of 0-order) | |
| Molecular distance edge | MDEN-22 (Xg1) | Molecular distance edge between all secondary nitrogens |
| MDEO-11 (Xg2) | Molecular distance edge between all primary oxygens | |
| MDEO-12 (Xg3) | Molecular distance edge between all primary and secondary oxygens | |
| MDEC-11 (Xg4) | Molecular distance edge between all primary carbons | |
| MLFERb | MLFER_BH (Xh) | Overall or summation solute hydrogen bond basicity |
| Path count | piPC3 (Xi1) | Conventional bond order ID number of order 3 (ln(1 + x)) |
| MPC9 (Xi2) | Molecular path count of order 9 | |
| Topological charge | GGI5 (Xj1) | Topological charge index of order 5 |
| GGI8 (Xj2) | Topological charge index of order 8 | |
| CPSAc | RPCS (Xk1) | Relative positive charge surface area |
| PNSA-1 (Xk2) | Partial negative surface area (sum of surface area on negative parts of molecule) | |
| Gravitational index | GRAV-4 (Xl) | Gravitational index of all pairs of atoms (not just bonded pairs) |
| WHIMd | Du (Xm1) | D total accessibility index (unweighted) |
| E1v (Xm2) | The first component accessibility directional WHIM index weighted by relative van der Waals volumes | |
The descriptors used and their interactions in polynomial terms are depicted in Fig. 7, in order to simplify the analysis of their contribution. The 2D model (Fig. 7a) uses 11 descriptors from 6 different groups, the most abundant being the molecular distance edge descriptors, which are topological descriptors that describe structural differences between compounds.56 The MM model (Fig. 7b) utilizes 13 descriptors from 10 groups, three of them being 3D descriptors. Similar can be observed in the case of SE model (Fig. 7c): 16 descriptors were selected from 10 groups, whereby three of them are 3D descriptors. A significant number of descriptors (nine) are shared between SE and MM model, while the same five 2D descriptors are common for all three models. These five descriptors (MDEO-11, MDEN-22, MLFER_BH, GGI5 and GGI8) can be found in the standalone terms of different degrees and in combined polynomial terms in which they describe synergetic effect on transition temperature.
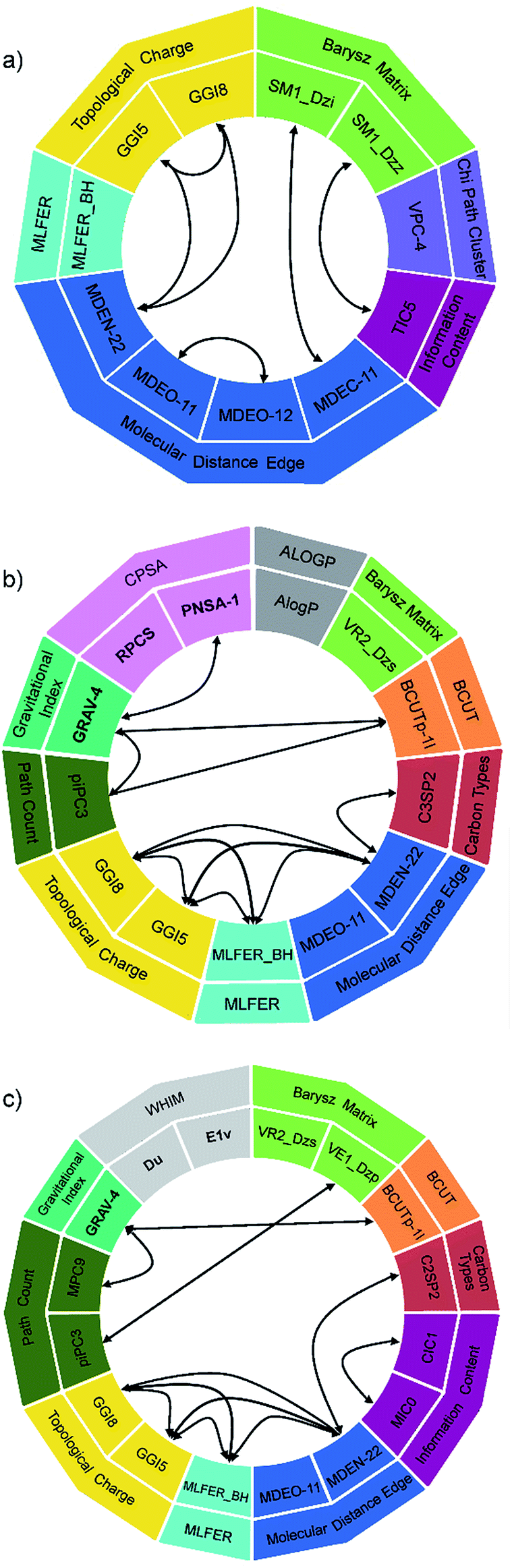 | ||
| Fig. 7 Descriptors and their interactions: (a) 2D, (b) MM and (c) SE model. | ||
Concerning the complexity of GMDH equations, it should be emphasized that for the majority of descriptors the assessment of their contribution can be performed only if synergetic effect is taken into account.
Also, about half of the descriptors are based on graph theory and similar mathematics, and therefore are difficult to interpret.57 In order to decode the impact of descriptors on the transition temperature, GMDH models need to be split on several sub-equations according to the descriptors interactions (coloured terms in eqn (8)–(10)).
Molecular distance edge (MDE) descriptors can be directly linked to the molecular structure. The MDEO-11 gives the distance between all primary oxygen atoms, while the MDEO-12 accounts for the distance between all primary and secondary oxygen atoms. Concerning that in this study MDEO-11 and MDEO-12 describe a similar structural feature (mainly the number of ester groups and their position and orientation), MDEO-11 was used in all three models, while MDEO-12 was present only in the 2D model (in the combined polynomial term with MDEO-11). From eqn (9) and (10) it can be observed that MDEO-11 individually contributes negatively to the transition temperatures of LCs, while in the 2D model (eqn (8)) its synergetic effect with MDEO-12 is prevalent, thus this negative influence is determined by the position of primary and secondary oxygen atoms.
In this study, MDEN-22 encodes information about the number and position of azo and azomethine groups, and it also can be found in all three models, whereby it affects transition temperature synergistically with several other descriptors (topological charge, carbon types and MLFER). Since the distribution of charge determines the nature of intermolecular forces,58 the selected topological charge descriptors (GGI5 and GGI8) suggest that the net charge transfer between five and eight atoms, among others, mostly affects the transition temperature. Carbon type descriptors, namely C3SP2 and C2SP2, indicate the type of linkage groups and presence of substituents on the phenyl rings. MLFER_BH is a measure of all hydrogen bond acceptor sites of a molecule, thus it describes the ability of molecule to form hydrogen bonds, which has influence on the transition temperature.
MDEC-11 descriptor affects transition temperature synergistically with Barysz matrix spectral moment descriptor. It decodes the influence of the size of molecule on the transition temperature.
GRAV-4 is the only 3D descriptor that is common for both 2 & 3D models, and it synergistically affects the transition temperature with BCUT, CPSA and path count descriptors. The gravitational index simultaneously gives the atomic masses and their distribution in a molecule, and it was found that it reflects most adequately molecular size-dependent bulk effects on the boiling points.59
Conclusion
In this study, nonlinear GMDH-type QSPR models were developed to predict transition temperatures for a dataset of 243 five-ring bent-core LC compounds, using multi-filter feature selection approach based on chi square and v-WSH algorithm. Descriptors were selected from a pool of 2D, and two pools of 2D and 3D ones, optimized by molecular mechanics (MM) and semi-empirical (SE) method. The final subset selection was performed using GMDH algorithm during the learning process. The models were compared using Taylor diagram, and a detailed evaluation of their performance (external testing, outlier analysis and randomization) was performed. Although all models demonstrated good accuracy (R2 = 0.92), the MM has showed slightly better performance, with a RMSE of 6.52 K for the external test set. Concerning that GMDH-type neural network gives polynomial equation that describe relationship between output and selected inputs, the obtained models have allowed the identification of key structural features that alter the transition temperature of five ring bent-core LCs.Acknowledgements
The authors are grateful to the Ministry of Education, Science and Technological Development of the Republic of Serbia for financial support [project numbers 172007 and 172013].Notes and references
- P. J. Collings and M. Hird, Introduction to Liquid Crystals: Chemistry and Physics, Taylor & Francis Ltd., London, UK, 1997 Search PubMed.
- H. Takezoe and Y. Takanishi, Jpn. J. Appl. Phys., 2006, 45, 597–625 CrossRef CAS.
- A. Eremin and A. Jákli, Soft Matter, 2013, 9, 615–637 RSC.
- S. R. Johnson and P. C. Jurs, Chem. Mater., 1999, 11, 1007–1023 CrossRef CAS.
- J. Xu, L. Wang, H. Zhang, C. Yi and W. Xu, Mol. Simul., 2010, 36, 26–34 CrossRef CAS.
- Z. G. Gong, R. S. Zhang, B. B. Xia, R. J. Hu and B. T. Fan, QSAR Comb. Sci., 2008, 27, 1282–1290 CAS.
- J. Antanasijević, V. Pocajt, D. Antanasijević, N. Trišović and K. Fodor-Csorba, Liq. Cryst., 2016, 43, 1028–1037 CrossRef.
- J. H. Al-Fahemi, Liq. Cryst., 2014, 41, 1575–1582 CrossRef CAS.
- Y. Ren, H. Liu, X. Yao, M. Liu and B. Fan, Liq. Cryst., 2007, 34, 1291–1297 CrossRef CAS.
- J. Antanasijević, D. Antanasijević, V. Pocajt, N. Trišović and K. Fodor-Csorba, RSC Adv., 2016, 6, 18452–18464 RSC.
- M. Eklund, U. Norinder, S. Boyer and L. Carlsson, J. Chem. Inf. Model., 2014, 54, 837–843 CrossRef CAS PubMed.
- R. E. Abdel-Aal, J. Biomed. Inf., 2005, 38, 456–468 CrossRef CAS PubMed.
- A. P. Alves da Silva, V. H. Ferreira and R. M. G. Velasquez, Int. J. Forecast., 2008, 24, 616–629 CrossRef.
- I. Guyon and A. Elisseeff, J. Mach. Learn. Res., 2003, 3, 1157–1182 Search PubMed.
- H. Li, Z. Zhong, L. Li, R. Gao, J. Cui, T. Gao, L. H. Hu, Y. Lu, Z. M. Su and H. Li, J. Comput. Chem., 2015, 36, 1036–1046 CrossRef CAS PubMed.
- S. Sasikala, S. Appavu alias Balamurugan and S. Geetha, Applied Computing and Informatics, 2016, 12, 117–127 CrossRef.
- J. K. Wegner, H. Fröhlich and A. Zell, J. Chem. Inf. Comput. Sci., 2004, 44, 921–930 CrossRef CAS PubMed.
- S. A. Kalogirou, Prog. Energy Combust. Sci., 2003, 29, 515–566 CrossRef CAS.
- A. G. Ivakhnenko, IEEE Transactions on Systems, Man, and Cybernetics, 1971, SMC-1, 364–378 CrossRef.
- S. J. Farlow, Self-Organizing Methods in Modeling: GMDH Type Algorithms, Marcel Dekker, Inc., New York, 1984 Search PubMed.
- M. Rahimi, R. Beigzadeh, M. Parvizi and S. Eiamsa-ard, Heat Mass Transf., 2016, 52, 1585–1593 CrossRef CAS.
- A. G. Ivakhnenko and G. A. Ivakhnenko, Pattern Recogn. Image Anal., 1995, 5, 527–535 Search PubMed.
- T. Kondo, J. Ueno and S. Takao, Procedia Comput. Sci., 2013, 22, 172–181 CrossRef.
- M. Najafzadeh and S. Y. Lim, Earth Sci. Inform., 2015, 8, 187–196 CrossRef.
- M. Sheikholeslami, F. Bani Sheykholeslami, S. Khoshhal, H. Mola-Abasia, D. D. Ganji and H. B. Rokni, Neural Comput. Appl., 2014, 25, 171–178 CrossRef.
- I. Ebtehaj, H. Bonakdari, A. Hossein Zaji, H. Azimi and F. Khoshbin, Engineering Science and Technology, an International Journal, 2015, 18, 746–757 CrossRef.
- ChemAxon Ltd., Marvin, 2014 Search PubMed.
- Hypercube Inc., HyperChem8.0, 2007 Search PubMed.
- S. Ananda Rama Krishnan, W. Weissflog and R. Friedemann, Liq. Cryst., 2005, 32, 847–856 CrossRef.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- J. Xu, L. Wang, L. Wang, X. Shen and W. Xu, J. Comput. Chem., 2011, 32, 3241–3252 CrossRef CAS PubMed.
- D. Ballabio, V. Consonni, A. Mauri, M. Claeys-Bruno, M. Sergent and R. Todeschini, Chemom. Intell. Lab. Syst., 2014, 136, 147–154 CrossRef CAS.
- P. Ambure, R. B. Aher, A. Gajewicz, T. Puzyn and K. Roy, Chemom. Intell. Lab. Syst., 2015, 147, 1–13 CrossRef CAS.
- StatSoft. Inc., Statistica (data analysis software system), version 10 trial, Tulsa, USA, 2010 Search PubMed.
- D. Newby, A. A. Freitas and T. Ghafourian, J. Chem. Inf. Model., 2013, 53, 2730–2742 CrossRef CAS PubMed.
- S. J. Farlow, Am. Stat., 1981, 35, 210–215 Search PubMed.
- D. E. Scott and C. E. Hutchison, The GMDH Algorithm – A technigue for economic modelling, report No. ECE-SY-67–1, Massachussetts, 1976 Search PubMed.
- I. V. Tetko, T. I. Aksenova, V. V. Volkovich, T. N. Kasheva, D. V. Filipov, W. J. Welsh, D. J. Livingstone and A. E. P. Villa, SAR QSAR Environ. Res., 2000, 11, 263–280 CrossRef CAS PubMed.
- V. Puig, M. Witczak, F. Nejjari, J. Quevedo and J. Korbicz, Eng. Appl. Artif. Intell., 2007, 20, 886–897 CrossRef.
- M. Witczak, J. Korbicz, M. Mrugalski and R. J. Patton, Control Eng. Pract., 2006, 14, 671–683 CrossRef.
- M. Mrugalski, Int. J. Appl. Math. Comput. Sci., 2013, 23, 157–169 Search PubMed.
- M. Witczak, M. Mrugalski and J. Korbicz, Neural Process. Lett., 2015, 42, 71–87 CrossRef.
- Ward systems group Inc., Neuroshell 2 v4.2, 2008 Search PubMed.
- A. R. Barron, in Self-Organizing Methods in Modeling: GMDH Type Algorithms, ed. S. J. Farlow, Marcel Dekker, Inc., New York, 1984, vol. 54, pp. 87–103 Search PubMed.
- A. S. Ahmad, M. Y. Hassan, M. P. Abdullah, H. A. Rahman, F. Hussin, H. Abdullah and R. Saidur, Renewable Sustainable Energy Rev., 2014, 33, 102–109 CrossRef.
- K. E. Taylor, J. Geophys. Res., 2001, 106, 7183–7192 CrossRef.
- L. Chen, C. Chu, J. Lu, X. Kong, T. Huang and Y.-D. Cai, Mol. BioSyst., 2015, 11, 2541–2550 RSC.
- E. Pourbasheer, A. Banaei, R. Aalizadeh, M. R. Ganjali, P. Norouzi, J. Shadmanesh and C. Methenitis, J. Ind. Eng. Chem., 2015, 21, 1058–1067 CrossRef CAS.
- K. Roy and J. T. Leonard, J. Chem. Inf. Model., 2005, 45, 1352–1368 CrossRef CAS PubMed.
- S. S. So and M. Karplus, J. Med. Chem., 1997, 40, 4347–4359 CrossRef CAS PubMed.
- D. Wang, Y. Yuan, S. Duan, R. Liu, S. Gu, S. Zhao, L. Liu and J. Xu, Chemom. Intell. Lab. Syst., 2015, 143, 7–15 CrossRef CAS.
- J.-B. Wang, D.-S. Cao, M.-F. Zhu, Y.-H. Yun, N. Xiao and Y.-Z. Liang, J. Chemom., 2015, 29, 389–398 CrossRef CAS.
- C. Rücker, G. Rücker and M. Meringer, J. Chem. Inf. Model., 2007, 47, 2345–2357 CrossRef PubMed.
- M. Nekoeinia, S. Yousefinejad and A. Abdollahi-Dezaki, Ind. Eng. Chem. Res., 2015, 54, 12682–12689 CrossRef CAS.
- K. Roy, S. Kar and R. N. Das, in A Primer on QSAR/QSPR Modeling, Springer, New York, 2015, pp. 37–59 Search PubMed.
- L. Jiao, X. Wang, S. Bing, Z. Xue and H. Li, RSC Adv., 2015, 5, 6617–6624 RSC.
- K. Varmuza, P. Filzmoser and M. Dehmer, Comput. Struct. Biotechnol. J., 2013, 5, e201302007 CrossRef PubMed.
- J. Galvez, R. Garcia, M. T. Salabert and R. Soler, J. Chem. Inf. Comput. Sci., 1994, 34, 520–525 CrossRef CAS.
- A. R. Katritzky, L. Mu, V. S. Lobanov, M. Karelson and V. Gaines, J. Phys. Chem., 1996, 100, 10400–10407 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |