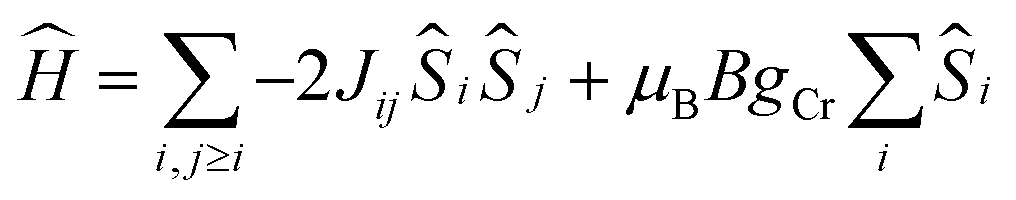DOI:
10.1039/C6RA14811E
(Paper)
RSC Adv., 2016,
6, 73668-73676
Solvothermal synthesis of discrete cages and extended networks comprising {Cr(III)3O(O2CR)3(oxime)3}2− (R = H, CH3, C(CH3)3, C14H9) building blocks†
Received
7th June 2016
, Accepted 21st July 2016
First published on 28th July 2016
Abstract
The synthesis, structural and magnetic characterisation of a family of related Cr(III) cages are reported. Each member comprises {Cr(III)3O(O2CR1)3(R2-sao)3}2− (R1 = H, CH3, C(CH3)3, C14H9; R2 = Me, Ph, tBu, C10H8) triangles linked by Na+ cations, resulting in either the discrete complexes [H3O][NEt4]2[NaCr(III)6O2(O2C-C14H9)6(Naphth-sao)6] (1) and [Na4Cr(III)6O2(O2CC(CH3)3)6(3,5-di-tBu-sao)6(MeCN)6] (3); or the extended networks [H3O]2[Na2Cr(III)6O2(O2CH)6(Ph-sao)6(MeCN)2(H2O)2]n·4MeCN (2); [H3O][Na3Cr(III)6O2(O2CCH3)6(Me-sao)6(MeCN)]n (4) and [Na2Cr(III)3O(O2CCH3)3(Me-sao)3(H2O)6]n·3MeCN (5). Magnetic susceptibility data obtained for 2 and 4 reveal weak antiferromagnetic exchange between the Cr(III) ions in the triangles.
Introduction
Hydro- or solvothermal synthesis comprises the heating of sealed reaction mixtures within Teflon-lined autoclaves. The high temperatures and pressures achieved can often lead to products that cannot be obtained under ambient conditions, nor via reflux or microwave heating.1 Controlled heating and cooling rates also aid crystallisation and can even be exploited to preferentially define crystal size and shape.2 In the coordination chemistry of large polymetallic cages of paramagnetic metal ions, the use of solvothermal methods has often promoted the formation of high symmetry species which are much sought after since they can display a range of fascinating and potentially useful low temperature physics.1,3
The synthesis of Cr(III) cages is dominated by its kinetic inertness and the need for high temperatures and pressures. Although several methodologies and structure types have been reported in the literature for many years,1,2,4 perhaps the most significant and well characterized complexes are the wheels of Winpenny and Timco, including elegant families of homo- (e.g. [Cr8],5 [Cr9]6 and [Cr10]7) and hetero-metallic ([Cr(III)7M1]; M = Ni(II), Co(II), Fe(II), Mn(II), Cd(II))8 ring structures, and their related horseshoes.9 These numerous and highly versatile architectures have also proven to be excellent models for molecular qubits with potential application in quantum information processing,10 as the macrocyclic element of a rotaxane based molecular shuttle,11 and more recently as building blocks in the self-assembly of larger premeditated multi-component assemblies.12
Building upon previous work concerning the tuning of the magnetic properties of salicyaldoxime-bridged [Mn3] and [Mn6] single-molecule magnets,13 we have now employed solvothermal synthesis in order to investigate the coordination chemistry of oxime-based ligands with Cr(III). This is an under-exploited area of chemistry, with a CSD search revealing just a handful of examples.14 Herein we report here the solvothermal synthesis and structural and magnetic characterisation of a family of discrete cages (1 and 3) and extended network materials (2, 4 and 5), each comprising trimetallic oxo-centred {Cr(III)3O(O2CR1)3(R2-sao)3}2− (R1 = H, CH3, C(CH3)3, C14H10; R2 = Me, tBu, Naphth, Ph) units linked via Na+ cations. Interestingly the structures of 1–5 are clearly rather different to the complexes [Cr(III)6O2(O2CR1)2(salox)6(H2O)2(R2CN)2] (R1 = CH3, C(CH3)3, C6H5; R2 = CH3, C3H7, C4H9; salox = salicylaldoxime), reported by Chaudhuri and co-workers, which are analogous to the aforementioned [Mn6] complexes.14d
Results and discussion
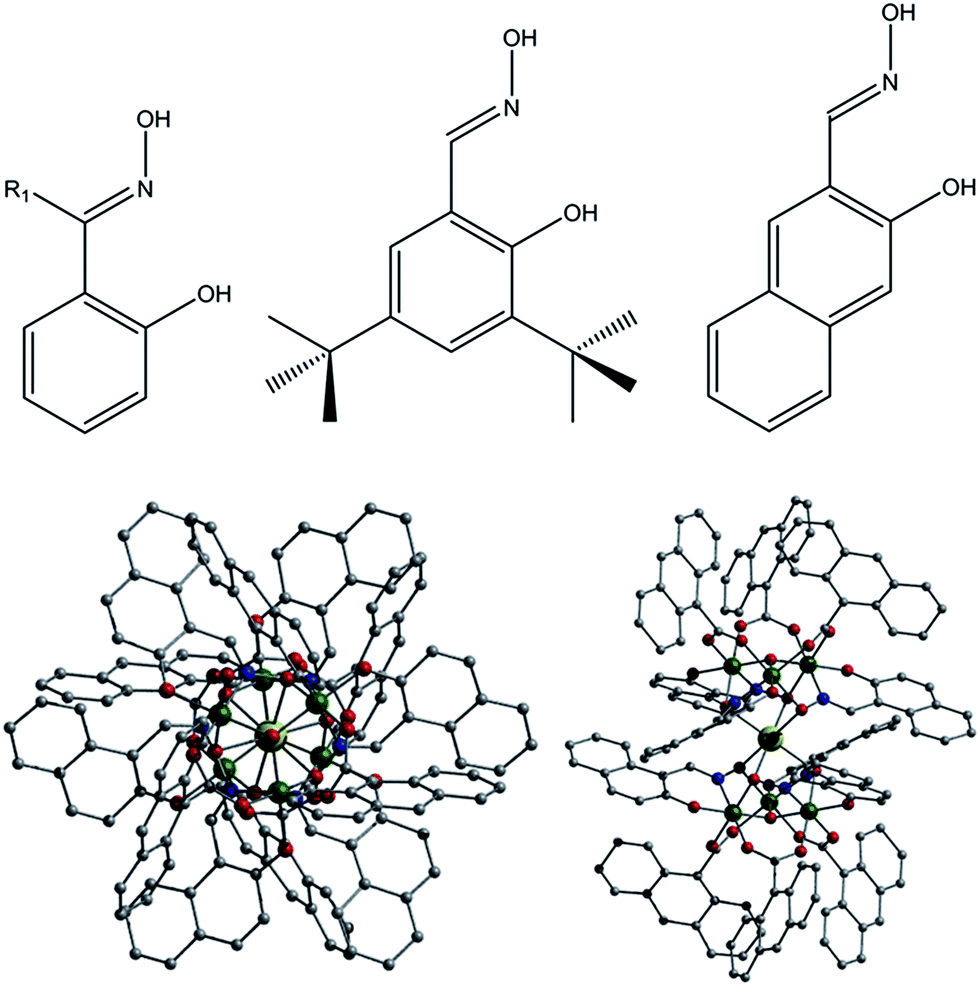
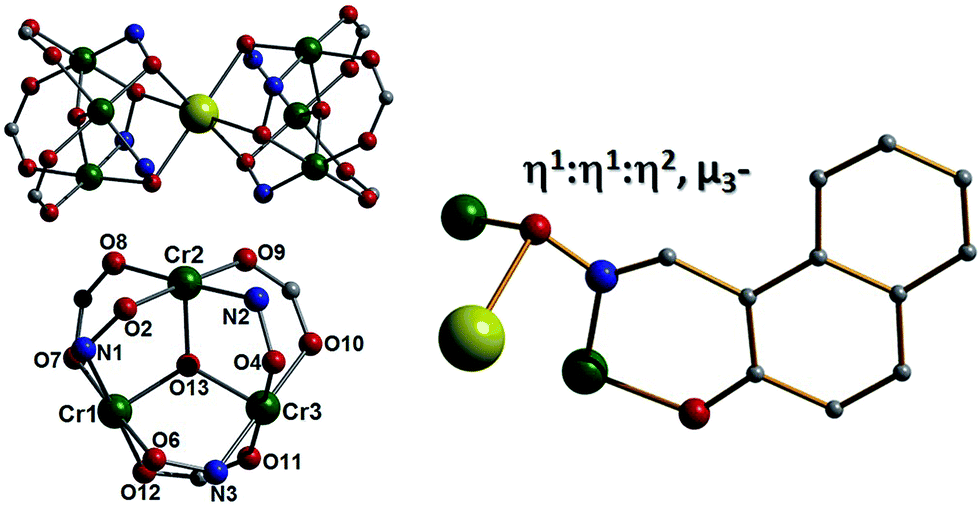
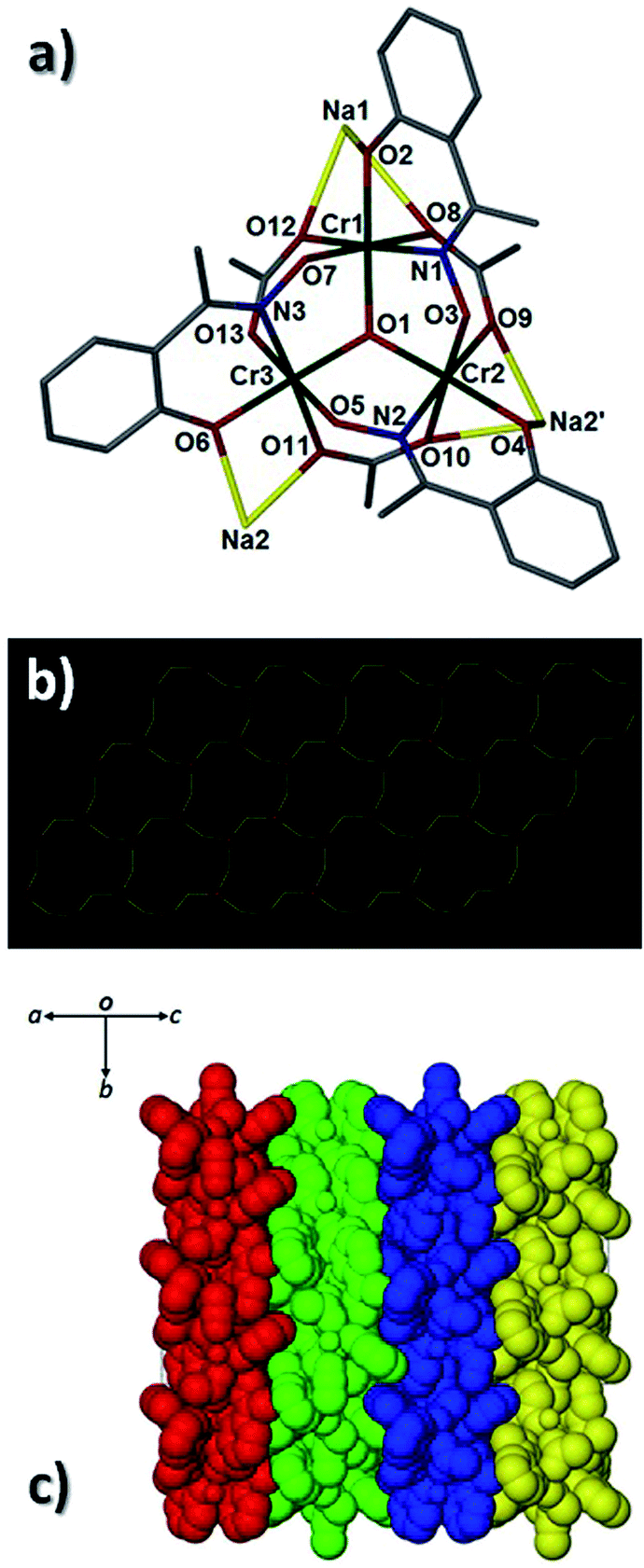
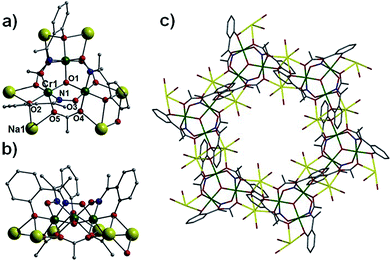
Pertinent crystallographic data for all five complexes are given in Tables S1 and S2.† All metal oxidation states were confirmed using bond valence sum (BVS) calculations and bond length and charge balancing considerations. The aerobic stirring of an acetonitrile solution of CrCl3·6H2O with equimolar amounts of Naphth-saoH2, sodium-9-anthracene carboxylate and base (NEt4[OH]) affords a green slurry. When this slurry is heated solvothermally for a period of 24 hours at T = 100 °C, followed by cooling at a rate of 3 °C per hour, crystals of the heptanuclear complex [H3O][NEt4]2[Na1Cr(III)6(O)2(O2C-C14H9)6(Naphth-sao)6] (1) are obtained. Longer cool down periods (range tested: 1–3 °C per hour) have no significant effect on yield and crystal quality. Complex 1 crystallises in the monoclinic space group C2/c, with one half [Na1Cr(III)6] unit in the asymmetric unit. The core in 1 has a sandwich-like shape where a central Na+ ion (Na1) lies at the midpoint of two {Cr(III)3O(O2C-C14H9)3(Naphth-sao)3}2− triangular units, which are related by an inversion centre lying at the Na+ position (Fig. 1). The carboxylate/oxime ligand distribution observed in these {Cr(III)3O(O2C-C14H9)3(Naphth-sao)3}2− moieties in 1 (and in 2–5) have been previously observed only in Mn(III) coordination chemistry,15 and are adaptations on the classic and ubiquitous trinuclear basic carboxylates [Cr3O(O2CR)6(L)3]0/+ (L = H2O, pyridine etc.),16 whereby the {Cr(III)3O}7+ core is maintained while substituting half the carboxylates for three Naphth-sao2− ligands to give the structure in 1 (Fig. 1). Each edge of these trinuclear fragments are bridged by one μ-O2C-anthracenoate (O2C-C14H9) and one η1:η1:η2, μ3-Naphth-sao2− ligand which occupy opposite sides of the {Cr(III)3O}7+ plane (Fig. 1 and 2). The O donor atoms of the carboxylates exclusively bridge the Cr(III) centres within the triangular units. The Naphth-sao2− ligands also bridge between neighbouring Cr ions, but also connect the {Cr(III)3O}7+ units to the central Na+ ion via six oximic O-atoms (O2, O4, O6 and symmetry equivalent, s.e.) with distances lying in the 2.377(5)–2.442(5) Å range (Fig. 2). The resultant [Na1Cr(III)6(O)2(O2C–C14H9)6(Naphth-sao)6]3− anionic clusters are charge balanced by a hydronium ion and two [NEt4]+ counter ions.
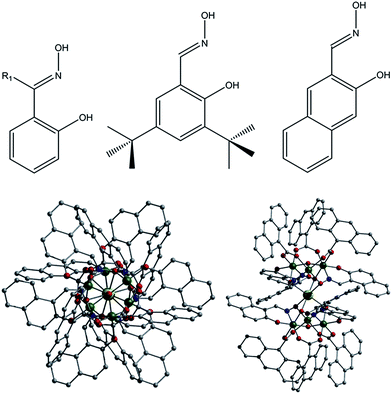 |
| | Fig. 1 (Top) ChemDraw representations of the derivatised salicyaldoxime ligands used in this work (where R1 = Me, Ph). (Bottom) Crystal structure of 1 as viewed perpendicular (left) and parallel (right) to the {Cr(III)3O}7+ planes. Colour code (used throughout this work): Cr (green), O (red), N (blue), Na (yellow), C (grey). All hydrogen atoms have been omitted for clarity. | |
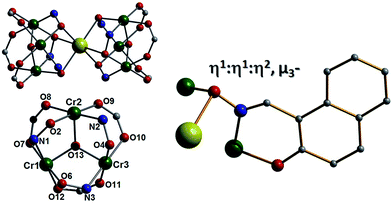 |
| | Fig. 2 (Top left) Core in 1 highlighting the major connection pathways between the two {Cr(III)3O}7+ triangular units and the central Na+ ion. (Bottom left) Core in 1 illustrating the oximic (Cr–N–O–Cr) and carboxylate (Cr–O–C–O–Cr) bridges within the {Cr(III)3O}7+ triangular units. (Right) Bonding mode exhibited by the Naphth-sao2− ligands in 1. | |
The [NEt4]+ counter anions in 1 sit within pockets formed by two neighbouring {NaCr6} units, and are held in position by numerous C–H⋯π interactions between ethyl protons and the surrounding aromatic rings of the Naphth-sao2− and −O2C–C14H9 ligands (e.g. C85(H85B)⋯[C15–C20]centroid = 2.646 Å and C83(H83B)⋯[C66–C71]centroid = 2.803 Å; Fig. S1†). The {NaCr6} units arrange into 2D brickwork sheets (bc plane) which are stacked in parallel rows along the a unit cell axis (Fig. S2†).
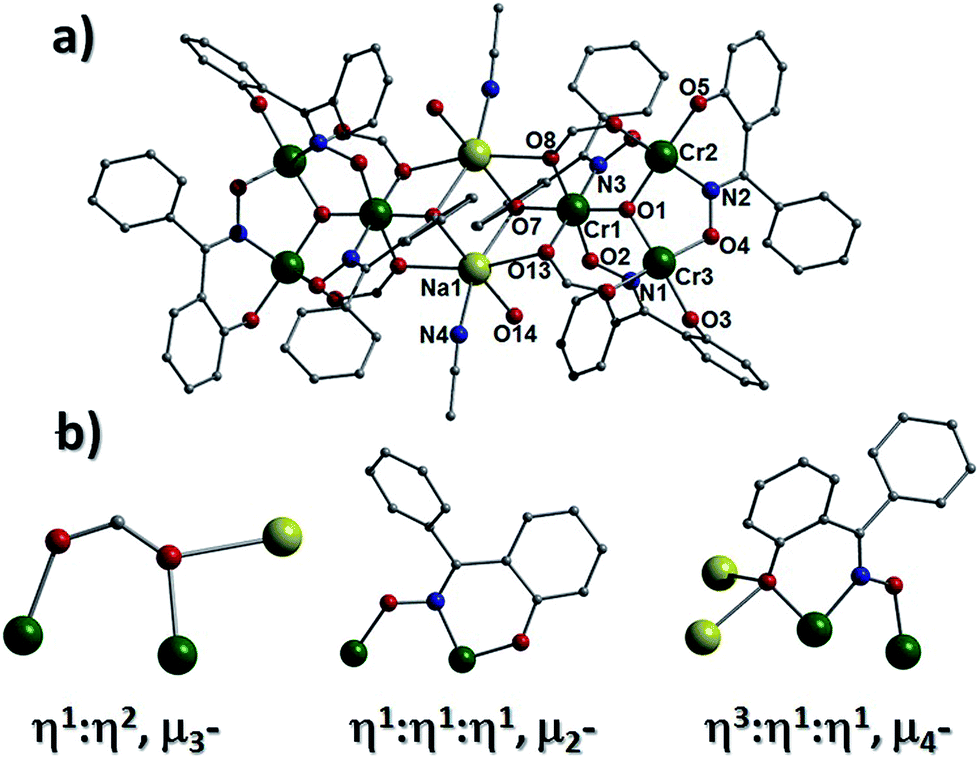
The solvothermal reaction of CrCl3·6H2O, Ph-saoH2, sodium formate and tetraethylammonium hydroxide in MeCN, affords the extended network [H3O]2[Na2Cr(III)6O2(O2CH)6(Ph-sao)6(MeCN)2(H2O)2]n·4MeCN (2) (Fig. 3). Complex 2 crystallises in the monoclinic space group P21/n and its core comprises two triangular [Cr(III)3O(O2CH)3(Ph-sao)3]2− units that, unlike in 1, are linked via two central distorted octahedral Na+ ions (Na1 and s.e.). An inversion centre lies at the midway point between the alkali metals. The coordination spheres at the two central Na+ centres are each completed by terminal H2O and MeCN ligands (Na1–N4 = 2.441(12) Å, Na1–O14 = 2.328(7) Å). The six carboxylates in 2 employ a combination of μ- and η1:η2, μ3-bonding motifs to fuse the central Na+ ions (Na1 and s.e.) to the {Cr(III)3O}7+ units, while also linking the Cr(III) centres within the triangular moieties. Similarly, the six Ph-sao2− ligands exhibit both η1:η1:η1 μ2- and η3:η1:η1 μ4-bonding modes to connect the Na+ ions to the [Cr(III)3] triangular units (Fig. 3b). Two symmetry equivalent hydronium ions (O15 and s.e.) lie juxtaposed to the core in 2 and are held in position through H-bonding interactions with O-donor atoms (O3, O9–O11) of nearby formate and Ph-sao2− ligands (O15⋯O3 = 2.718 Å, O15⋯O9 = 2.921 Å, O15⋯O10 = 2.835 Å and O15⋯O11 = 2.919 Å) and two MeCN solvent of crystallisation (N5 and N6) (O15⋯N5 = 3.077 Å, O15⋯N6 = 2.935 Å). In combination with the linker Na+ ions, these hydronium ions effectively connect the individual [Cr3] units in 2 towards the formation of wave-like 2D sheets that intersect the bc plane of the unit cell and stack upon one another along the a cell direction in a space efficient parallel fashion (Fig. 4b and S3†). The overall result is an [3,6] network topology in 2 as shown in Fig. 4a.
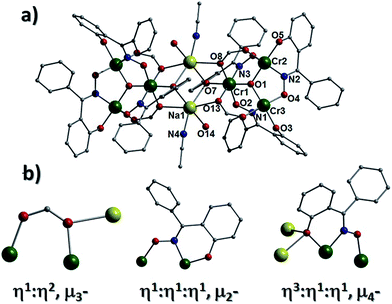 |
| | Fig. 3 (a) Crystal structure of 2 minus all hydrogen atoms. (b) Selection of bonding modes exhibited by the formate (left) and Ph-sao2− (middle and right) ligands in 2. | |
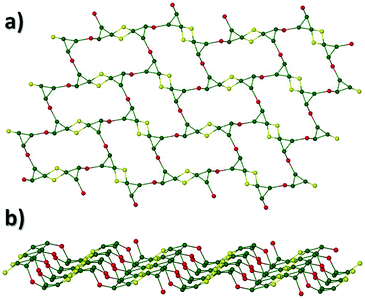 |
| | Fig. 4 (a) Connectivity schematic taken from the crystal data of 2 illustrating its [3,6] net topology. Colour code: green (Cr), yellow (Na) and red (O). Note: the yellow and red spheres represent Na1 and O15 (hydronium ion), respectively. (b) A single 2D sheet observed in 2. | |
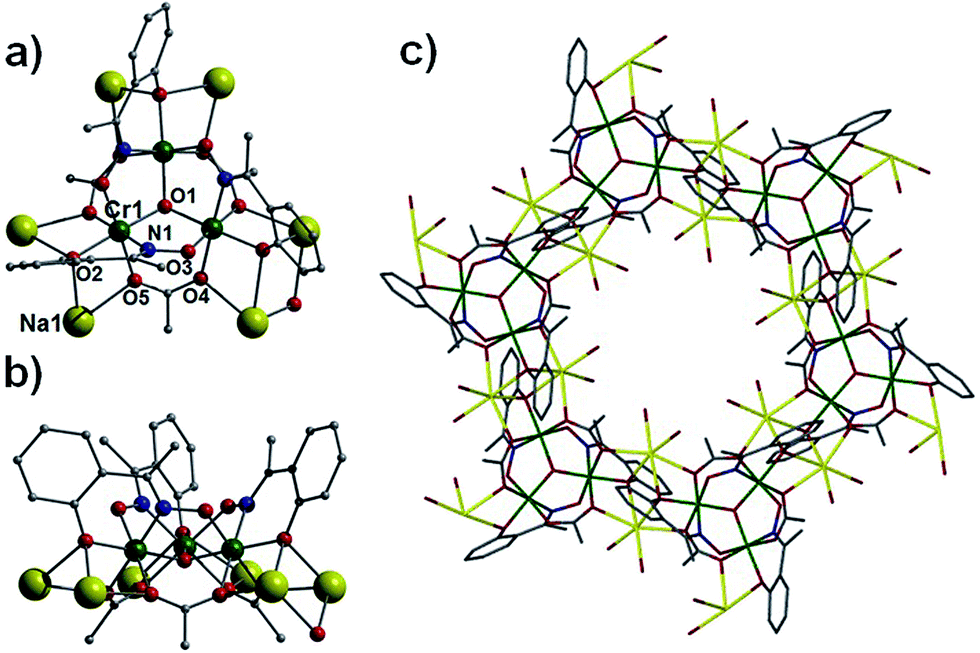
Employment of the ligand 3,5-di-tert-butyl-salicyaldoxime in combination with pivalate co-ligands in an analogous reaction to those described above gives rise to the complex [Na4Cr(III)6(O)2(O2CC(CH3)3)6(3,5-di-tBu-sao)6(MeCN)6] (3). Complex 3 crystallises in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c and bears a similar sandwich-like core to that observed in 1 (compare Fig. 5c with 2). However in this particular case the two {Cr(III)3O(O2CC(CH3)3)3(3,5-di-tBu-sao)3}2− moieties are connected through a belt of four Na+ ions (Na1–2 and s.e.). Na2 (as in 1) is bound by oximic O-atoms (O2, O6 and s.e.) and lies at the centre of the cage, equidistant between the two central O2− ions (O5 and O7) located at the centre of each {Cr(III)3O}7+ unit. The three symmetry related sodium centres (Na1 and s.e.) are situated in the same plane as the central Na2 and together form a star shaped arrangement (Fig. 5a). The coordination spheres at Na1 (and s.e.) are completed by two terminal MeCN ligands (Na1–N2 = 2.497(7) Å, Na1–N3 = 2.454(7) Å) (Fig. 5b and d). As observed in 1, the carboxylates in 3 bridge only the Cr(III) ions within each trinuclear unit using the common μ-bridging mode. As a result the tBu-sao2− ligands must assume the role of connecting the {Cr(III)3O}7+ units to the central belt of Na+ ions and this is achieved using a η2:η1:η3, μ5-bonding motif (Fig. S4†). On moving from complex 1 to 3, it becomes apparent that the number of Na+ ions increases from one (in 1) to four (in 3); this may tentatively be assigned to the presence of less sterically demanding organic ligands. In the crystal of 3 the [Na4Cr6] moieties stack on top of one another resulting in a unit cell possessing aesthetically pleasing pseudo-superimposable 1D columns, with each unit linked by a 120° rotation (Fig. 6).
c and bears a similar sandwich-like core to that observed in 1 (compare Fig. 5c with 2). However in this particular case the two {Cr(III)3O(O2CC(CH3)3)3(3,5-di-tBu-sao)3}2− moieties are connected through a belt of four Na+ ions (Na1–2 and s.e.). Na2 (as in 1) is bound by oximic O-atoms (O2, O6 and s.e.) and lies at the centre of the cage, equidistant between the two central O2− ions (O5 and O7) located at the centre of each {Cr(III)3O}7+ unit. The three symmetry related sodium centres (Na1 and s.e.) are situated in the same plane as the central Na2 and together form a star shaped arrangement (Fig. 5a). The coordination spheres at Na1 (and s.e.) are completed by two terminal MeCN ligands (Na1–N2 = 2.497(7) Å, Na1–N3 = 2.454(7) Å) (Fig. 5b and d). As observed in 1, the carboxylates in 3 bridge only the Cr(III) ions within each trinuclear unit using the common μ-bridging mode. As a result the tBu-sao2− ligands must assume the role of connecting the {Cr(III)3O}7+ units to the central belt of Na+ ions and this is achieved using a η2:η1:η3, μ5-bonding motif (Fig. S4†). On moving from complex 1 to 3, it becomes apparent that the number of Na+ ions increases from one (in 1) to four (in 3); this may tentatively be assigned to the presence of less sterically demanding organic ligands. In the crystal of 3 the [Na4Cr6] moieties stack on top of one another resulting in a unit cell possessing aesthetically pleasing pseudo-superimposable 1D columns, with each unit linked by a 120° rotation (Fig. 6).
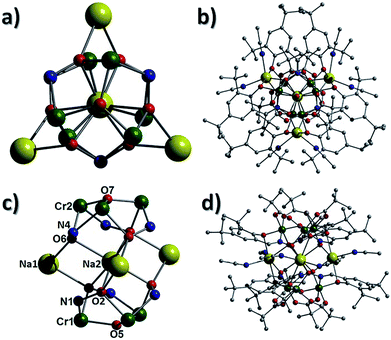 |
| | Fig. 5 Inorganic core (left) and complete cage (right) in 3, as viewed perpendicular (a and b) and parallel (c and d) to the {Cr(III)3O(O2CC(CH3)3)3(3,5-di-tBu-sao)3}2− triangular units. | |
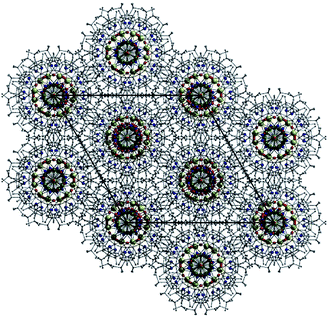 |
| | Fig. 6 Crystal structure of the hexagonal packing arrangement in 3 as viewed along the c axis of the unit cell. Hydrogen atoms have been omitted for clarity. | |
A combination of the Me-saoH2, acetate ligands and NaOH leads to the formation of [H3O][Na3Cr(III)6(O)2(O2CCH3)6(Me-sao)6(MeCN)]n (4) (Fig. 7). Complex 4 crystallises in the monoclinic I2/a space group and is the second example of an extended network in this family of compounds. Its structure comprises {Cr(III)3(O)(O2CCH3)3(Me-sao)3}2− nodes linked into a 2D [3,6] sheet-like topology via charge balancing Na+ ions. The connectivity within each {Cr(III)3O}7+ node is as follows: the three Me-sao2− ligands located within each [Cr3] moiety exhibit the η2:η1:η1, μ3-bonding mode and each span an edge of a trinuclear unit while also providing a connection to the Na+ ion nodes (Na1–2 and s.e.). An identical role is adopted by the three acetate ligands within each {Cr(III)3O}7+ unit in 4, using a combination of η2:η1, μ3- and η2:η2, μ4-bonding motifs (Fig. S5†). As a result the central {Cr(III)3O}7+ nodes are connected into 2D sheets via three bridging Na+ ions, resulting in a [3,6] topology as shown in Fig. 7b. The hydronium ions (O14 and s.e.) sit directly above the [Cr3] units in 4 and stitch together the individual 2D sheets through H-bonding interactions (e.g. O14⋯05 = 2.873 Å and O14⋯07 = 2.901 Å). The extended connectivity in 4 is also enhanced by numerous inter-sheet C–H⋯π interactions between aromatic oxime and neighbouring acetate protons (C24(H24B)⋯[C9–C14]centroid = 3.381 Å and C20(H20)⋯[C1–C6]centroid = 2.796 Å). Na1 exhibits a distorted octahedral geometry while Na2 possesses a distorted square-based pyramidal configuration (τ = 0.10).17 A single acetonitrile ligand completes the coordination sphere at Na1 (Na1–N4 = 2.437(7) Å).
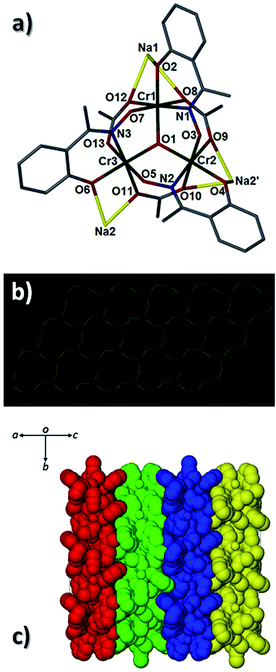 |
| | Fig. 7 (a) A single {Cr(III)3O(O2CCH3)3(Me-sao)3}2− node surrounded by three connecting Na+ ions as viewed perpendicular to the {Cr(III)3O}7+ plane in 4. (b) The 2D [3,6] network in 4 where the red vertices represent the O (O1) atom at the centre of the {Cr3O}7+ triangular nodes and the yellow vertices represent the bridging Na+ ions (Na1–2). (c) Colour coded and space-fill represented 2D nets in the unit cell of 4. | |
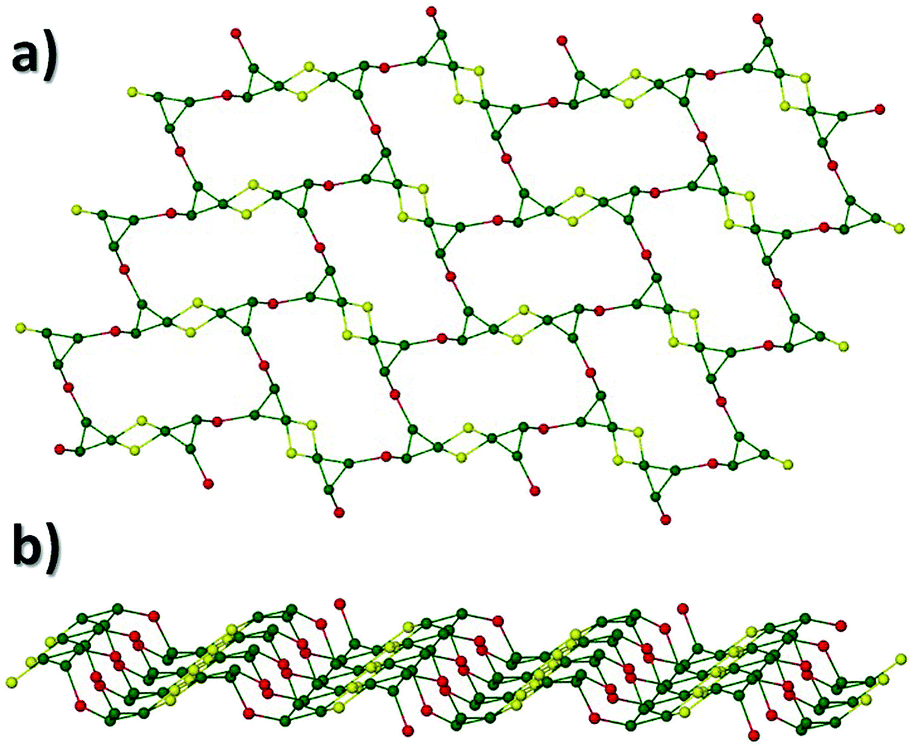
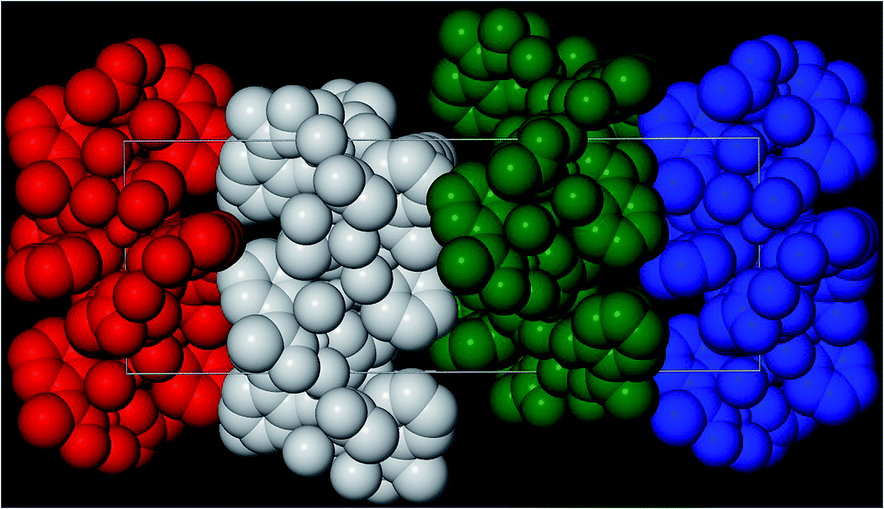
A repeat of the reaction that produced 4, but under ambient conditions affords green crystals of the species [Na2Cr(III)3O(O2CCH3)3(Me-sao)3(H2O)6]n·3MeCN (5) after the methanolic slurry is taken to dryness and recrystallized from MeCN (trigonal R space group). Akin to 4, complex 5 also exhibits a 2D [3,6] extended network topology and a core comprising {Cr(III)3O(O2CCH3)3(Me-sao)3(H2O)6}2− nodes linked by Na+ ions (Na1 and s.e.) to give a highly symmetrical hexagonal honeycomb array (Fig. 8 and 9).18 To create this 2D topology the Me-sao2− and −O2CCH3 ligands each bridge four metal centres (2 × Cr(III) and 2 × Na+) by adopting η3:η1:η1, μ4- and η2:η2, μ4-bonding modes, respectively (Fig. S6†). Two terminal H2O ligands (O6 and O7) bond to each sodium linker ion (Na1–O6 = 2.372(3) Å, Na1–O7 = 2.373(4) Å) and in doing so lie within the hexagonal cavity while effectively completing their distorted octahedral geometries. The individual 2D sheets in 5 pack in a parallel fashion along the c direction of the unit cell as illustrated in Fig. 10 (inter-sheet distance: Na1⋯Na1′ = 12.272 Å). Moreover these individual sheets arrange in a staggered parallel arrangement in relation to one another and therefore no significant pores or channels are observed in 5. However, MeCN solvent of crystallisation occupy the spaces in-between each of the 2D sheets and do so by arranging themselves into symmetry related triads around the three fold axis of the cell (down c), while also H-bonding with terminal H2O ligands (N2⋯O6 = 2.845 Å) (Fig. 9).
 |
| | Fig. 8 A single {Cr(III)3O}7+ unit along with the linker Na+ ions (yellow spheres) as viewed perpendicular (a) and parallel (b) to the triangular plane in 5. (c) An array of six {Cr(III)3O(O2CCH3)3(Me-sao)3}2− units linked by Na+ ions to form the hexagonal [3,6] honeycomb topology in 5. | |
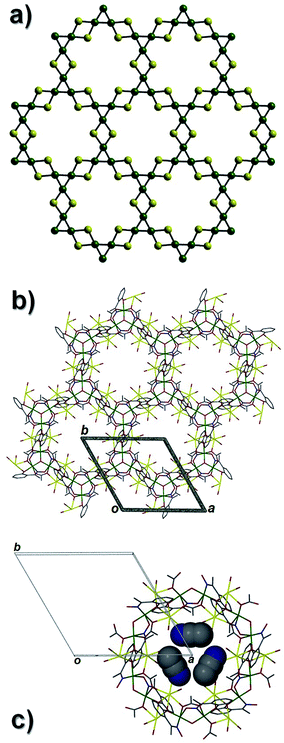 |
| | Fig. 9 (a) Connectivity schematic taken from the crystal data of 5 illustrating its [3,6] net topology (colour code as used previously). (b) The [3,6] 2D extended network in 5 as viewed along the c axis of the unit cell. Solvent molecules of crystallisation (MeCN) have been omitted for clarity. (c) Three symmetry equivalent MeCN solvents of crystallisation (space-fill representations) lying perpendicular to the three fold axis along c and reside in-between the individual 2D sheets in 5. | |
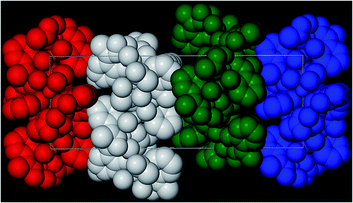 |
| | Fig. 10 Space-fill representation of the parallel stacking of the individual (colour coded) 2D [6,3] nets in 5, highlighting their staggered arrangements with respect to one another. All MeCN solvent molecules and H atoms have been omitted for clarity. | |
TGA-DSC measurements
The TG trace of [Na4Cr(III)6O2(O2CC(CH3)3)6(3,5-di-tBu-sao)6(MeCN)6] (3) shows two distinct mass loss regions, with the initial loss of 10.4% corresponding to all (7.5 per cage) coordinated and interstitial MeCN units (calculated as 11.1%). The second (and much steeper) mass drop occurs at ∼420 °C and is attributed to the sublimation of 3 (or its related decomposition products). The corresponding trace for [Na2Cr3O(O2CCH3)3(Me-sao)3(H2O)6]n·3MeCN (5) exhibits three mass loss regions. The initial loss (in the 50–100 °C range) of 18.1% is attributed to the removal of interstitial MeCN solvent molecules and terminal H2O solvent ligands (calculated at 21.3%). The closely related second and third steps between 300 and 475 °C represent an 18% loss and is tentatively attributed to acetate ligand loss (calculated as 16.3%), which is rapidly followed by further ligand loss and eventual decomposition of 5 (Fig. S7†).
Magnetic susceptibility measurements
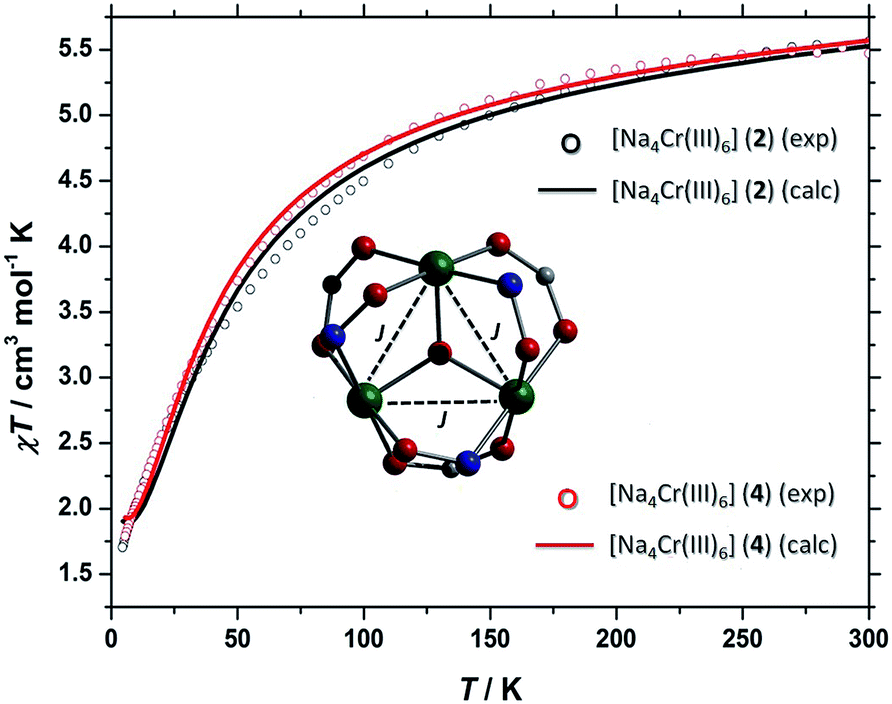
The d.c. molar magnetic susceptibility, χ, of polycrystalline samples of 2 and 4 were measured in an applied magnetic field, B, of 0.1 T, over the 5–300 K temperature, T, range (Fig. 11, where χ = M/B, and M is the magnetisation). Both show very similar behaviour, and we discuss the data per {Cr(III)3} triangle assuming no interaction through the Na+ ions. At room temperature, the χT products of both are approximately 5.5 cm3 mol−1 K, close to the value expected from the spin-only contribution to the magnetism of a trinuclear Cr(III) unit (5.6 cm3 mol−1 K, with gCr = 2.00). Upon cooling, the χT products decrease continuously to reach a value of approximately 1.8 cm3 mol−1 K per [Cr(III)3] at 5 K. This behaviour is indicative of weak antiferromagnetic interactions between the Cr(III) ions. For the interpretation of the magnetic properties of 2 and 4, we consider that they arise from an equilateral triangle with just one exchange interaction (Fig. 11-inset). Thus we used spin-Hamiltonian (1):| |
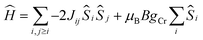 | (1) |
with i, j running over all Cr(III) centres of a [Cr(III)3] unit, J the isotropic magnetic exchange interaction between Cr(III) centres, Ŝ a spin-operator, μB the Bohr magneton, B the applied magnetic field, and gCr = 2, the isotropic g-factor common to all Cr(III) centres. The experimental χT data were numerically fitted to spin-Hamiltonian (1) by use of the simplex algorithm to afford the best-fit parameters J = −4.5 cm−1 for 2 and J = −4.0 cm−1 for 4. The best-fit curves are shown as solid lines in Fig. 11. It should be noted here that although the [Cr3] triangles are scalene in nature and thus the Cr(III) ions are not symmetry equivalent, employing a model with two or more exchange interaction parameters produces physically meaningless parameters, while also rendering any assessment of Na+ mediated inter-triangle exchange futile.
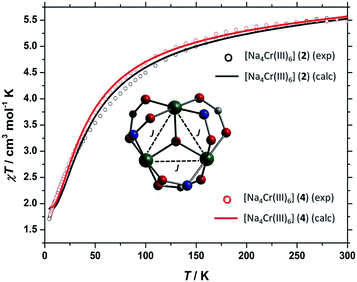 |
| | Fig. 11 χMT (per [Cr(III)3] triangle) versus T for polycrystalline samples of 2 and 4 taken in the T = 300–5 K temperature range in an applied field of B = 0.1 T. The solid lines are a fit of the experimental data to spin-Hamiltonian (1). | |
Concluding remarks
The solvothermal heating of Cr(III) salts in the presence of bulky R-saoH2 oxime ligands (Naphth-saoH2 and Ph-saoH2) with carboxylate co-ligands of varying sizes produces a series of structurally related discrete cages each comprising Na+ linked {Cr(III)3O(O2CR1)3(R2-sao)3}2− units. Siblings 1–5 are thus new members of the very small family of Cr(III) cages stabilised with phenolic oximes, and their formation suggests many more family members await discovery through judicious ligand design and the exploration of a variety of reaction conditions. Moreover, compounds 2, 4 and 5 are the first examples of extended network materials comprising Cr-oxime building blocks. Magnetic susceptibility measurements on 2 and 4 reveal weak antiferromagnetic exchange between neighbouring Cr(III) ions within the isolated triangular units.
Experimental section
Physical measurements
Infra-red spectra were recorded on a Perkin Elmer FT-IR Spectrum One spectrometer equipped with a Universal ATR Sampling accessory (NUI Galway). Elemental analysis were carried out by Marion Vignoles of the School of Chemistry microanalysis service at NUI Galway. Variable-temperature, solid-state direct current (d.c.) magnetic susceptibility data down to 5 K were collected on a Quantum Design MPMS-XL SQUID magnetometer equipped with a 7 T dc magnet (University of Edinburgh). Diamagnetic corrections were applied to the observed paramagnetic susceptibilities using Pascal's constants. All measured complexes were set in eicosane to avoid torqueing of the crystallites. All magnetic samples are collected as single-crystalline products and analysed using microanalysis and IR measurements prior to their magnetic assessment. If necessary, phase purity between cross-batches are validated using unit cell checks and IR measurements. TGA measurements were carried out by Dermot McGrath (NUI Galway) using a Rheometric Scientific STA 625.
X-ray crystallography
The structures of 1–5 were collected on an Xcalibur S single crystal diffractometer (Oxford Diffraction) using an enhanced Mo source. Each data reduction was carried out on the CrysAlisPro software package. The crystal structures of 1 and 3 was solved by an dual-space algorithm using SHELXT,19 whereas 2, 4 and 5 were solved by direct methods using SHELXS-97 (ref. 20) and refined by full matrix least squares using SHELXL-2014 (ref. 19) within OLEX2 (ref. 21) (structures 1 and 3) and SHELXL-97 (ref. 20) (structures 2, 4 and 5) within OSCAIL software package.22 All hydrogen atoms were placed in calculated positions. All non-hydrogen atoms were refined as anisotropic. Despite numerous attempts each single crystal data set obtained from 1–3 were found to consistently diffract poorly at higher angles. Our best data sets have been supplied in this work. Residual electron densities in solvent accessible voids and channels were observed in 1, 3 and 5 and so were modelled using the SQUEEZE program.23 The two large channels (volumes ∼ 970 Å3) in 1 contained extremely diffuse electron density and were assumed to contain numerous solvents of crystallisation (H2O and MeCN) along with the required oxonium charge balancing cation. CHN analysis on 1 supported these observations. Due to poor quality of data, SIMU, DELU and RIGU restraints were applied to the crystal structure in 1. Two tert-butyl groups in 3 (at C5 and C37, respectively) were each modelled as disordered over two sites with ratios of 61.7/38.3 (at C5) and 86.5/13.5 (at C37). Bond length (C–C) and angle (C–C–C) restraints were also required to maintain sensible geometries at these sites. Likewise, global SIMU/DELU and RIGY restraints were employed to improve the atomic displacement parameters at the disordered C6 and C6A sites (see cif for full details).
Materials and syntheses
All solvothermal reactions (1–4) were carried out in a Hereaus (UT6420-Thermo Scientific) oven using spring loaded stainless steel digestion vessels (23 cm3 capacity) produced by the Parr Instrument Company. All reagents and solvents were used as purchased. The salicyaldoxime ligands were synthesised by the reaction of the precursor ketone with hydroxylamine and sodium acetate in ethanol, as described in the literature.24 The synthesis of chromium pivalate was adapted from established methods,25 while chromium acetate was purchased from Sigma Aldrich and used without purification. Complex 5 was carried out on the bench top under ambient conditions using solvents that were used as purchased without further purification.
[H3O][NEt4]2[NaCr(III)6(O)2(O2C-C14H9)6(Naphth-sao)6] (1). CrCl3·6H2O (0.1 g, 0.37 mmol), Naphth-saoH2 (0.07 g, 0.037 mmol), sodium-9-anthracene carboxylate (0.09 g, 0.037 mmol) and tetraethylammonium hydroxide (0.2 cm3, 0.15 mmol) were stirred in MeCN (10 cm3) for 6 hours. This slurry solution was then transferred to a 23 cm3 capacity Teflon vial which was then lodged in a spring loaded stainless steel digestion vessel and placed in an oven. The oven temperature was raised to 100 °C over a period of 5 minutes and held at this temperature for 24 hours. The temperature was then lowered gradually down to room temperature over a period of 24 hours. Dark green crystals of 1 were collected and air dried to give a yield of 11%. Elemental analysis (%) calculated (found) for C172H193N8O54Na1Cr6 (1·H3O·27H2O): C 57.85 (58.14), H 5.45 (5.13), N 3.14 (3.28). FT-IR (cm−1): 3046(w), 1613(m), 1596(m), 1559(s), 1503(m), 1487(w), 1457(m), 1427(s), 1383(s), 1322(s), 1276(m), 1249(m), 1181(m), 1141(w), 1087(w), 1041(m), 1025(m), 951(s), 885(w), 864(w), 822(m), 785(m), 750(m), 735(s), 678(s).
[H3O]2[Na2Cr(III)6(O)2(O2CH)6(Ph-sao)6(MeCN)2(H2O)2]n·4MeCN (2). CrCl3·6H2O (0.1 g, 0.38 mmol), Ph-saoH2 (0.08 g, 0.38 mmol), sodium formate (0.05 g, 0.74 mmol) and tetraethylammonium hydroxide (0.2 cm3, 0.15 mmol) were stirred in MeCN (10 cm3) for 2 hours. This slurry solution was then transferred to a 23 cm3 capacity Teflon vial which was then placed in an spring loaded stainless steel digestion vessel and placed in an oven. The oven temperature was raised to 100 °C over a period of 5 minutes and held at this temperature for 24 hours. The temperature was then lowered gradually down to room temperature over a period of 24 hours. Dark green rhombic shaped crystals of 2 were collected and air dried to give a yield of 10%. Elemental analysis (%) calculated (found) for C96H88N12O30Na2Cr6: C 51.30 (51.01), H 3.95 (3.75), N 7.48 (7.76). FT-IR (cm−1): 3369(b), 1576(s), 1490(w), 1471(w), 1436(m), 1394(w), 1359(m), 1311(s), 1250(w), 1155(w), 1045(m), 1026(m), 957(s), 846(w), 759(s), 673(s).
[Na4Cr(III)6(O)2(O2CC(CH3)3)6(3,5-di-tBu-sao)6(MeCN)6] (3). Chromium pivalate (0.1 g, 0.20 mmol), 3,5-di-tert-butyl-saoH2 (0.15 g, 0.60 mmol) and NaOH (0.03 g, 0.75 mmol) were stirred in MeCN (10 cm3) for 2 hours. This slurry solution was then transferred to a 23 cm3 capacity Teflon vial which was then placed in an spring loaded stainless steel digestion vessel and then placed in an oven. The oven temperature was raised to 100 °C over a period of 5 minutes and held at this temperature for 24 hours. The temperature was then lowered gradually down to room temperature over a period of 24 hours. Dark green rhombic shaped crystals of 3 were collected and air dried to give a yield of 12%. Elemental analysis (%) calculated (found) for C132H198N12O26Na4Cr6 (3): C 57.17 (57.01), H 7.20 (7.45), N 6.06 (6.26). FT-IR (cm−1): 3406(vb), 2957(w), 1589(w), 1549(s), 1484(m), 1460(w), 1420(s), 1378(m), 1361(m), 1296(w), 1277(w), 1255(m), 1229(m), 1201(w), 1169(m), 1020(m), 987(w), 956(w), 898(w), 872(w), 835(m), 812(w), 788(m), 746(w), 715(s), 695(m).
[H3O][Na3Cr(III)6(O)2(O2CCH3)6(Me-sao)6(MeCN)]n (4). Chromium acetate (0.1 g, 0.17 mmol), Me-saoH2 (0.08 g, 0.53 mmol) and NaOH (0.03 g, 0.75 mmol) were stirred in MeCN (10 cm3) for 1.5 hours. This slurry solution was then transferred to a 23 cm3 capacity Teflon vial which was then placed in an spring loaded stainless steel digestion vessel and then placed inside an oven. The oven temperature was raised to 100 °C over a period of 5 minutes and held at this temperature for 24 hours. The temperature was then lowered gradually down to room temperature over a period of 24 hours. Dark green rhombic shaped crystals of 4 were collected and air dried to give a yield of 15%. Elemental analysis (%) calculated (found) for C62H72O30N7Na3Cr6 (4·3H2O): C 41.92 (41.58), H 4.09 (4.43), N 5.52 (5.75). FT-IR (cm−1): 3361(w), 2989(w), 2216(w), 1615(w), 1595(m), 1536(m), 1506(m), 1455(m), 1427(s), 1379(s), 1329(s), 1292(s), 1248(m), 1184(s), 1144(m), 1091(w), 1044 (s), 953(s), 881(w), 859(w), 824(s), 787(s), 777(s), 743(s).
[Na2Cr(III)3O(O2CCH3)3(Me-sao)3(H2O)6]n·3MeCN (5). Chromium acetate (0.25 g, 0.41 mmol), Me-saoH2 (0.19 g, 1.25 mmol) and NaOH (0.066 g, 1.65 mmol) were dissolved in MeOH (30 cm3) and stirred for 4.5 h. The solvent was then removed under reduced pressure and the green solid redissolved in MeCN. The resultant solution was filtered, and X-ray quality crystals of 5 were obtained upon slow evaporation of the mother liquor in 10% yield after 6 days. Elemental analysis (%) calculated (found) for C30H54O25N3Na2Cr3 (5·6H2O): C 34.03 (33.85), H 5.14 (4.64), N 3.96 (3.79). FT-IR (cm−1): 3357(b), 2167(w), 1581(m), 1550(vs), 1481(w), 1430(vs), 1301(vs), 1244(s), 1164(w), 1138(s), 1054(s), 1020(m), 971(m), 860(m), 754(s), 706(s), 949(vs).
Acknowledgements
The author (LFJ) would like to thank the SFI Investigator Program for funding (Investigator Project Number: 12/IP/1322). We would also like to thank the IRCSET Embark Fellowship Program for funding EH.
Notes and references
-
(a) R. Shaw, R. H. Laye, L. F. Jones, D. M. Low, C. Talbot-Eeckelaers, Q. Wei, C. J. Milios, S. Teat, M. Helliwell, J. Raftery, M. Evangelisti, M. Affronte, D. Collison, E. K. Brechin and E. J. L. McInnes, Inorg. Chem., 2007, 46, 4968 CrossRef CAS PubMed;
(b) D. M. Low, L. F. Jones, A. Bell, E. K. Brechin, T. Mallah, E. Riviére, S. J. Teat and E. J. L. McInnes, Angew. Chem., Int. Ed., 2003, 42, 3781 CrossRef CAS PubMed.
-
(a) N. Stock and S. Biswas, Chem. Soc. Rev., 2011, 112, 933 CrossRef PubMed;
(b) R. I. Walton, Chem. Soc. Rev., 2002, 31, 230 RSC.
-
(a) Y. Hou, X. Fang, K. D. Kwon, L. J. Criscenti, D. Davis, T. Lambert and M. Nyman, Eur. J. Inorg. Chem., 2013, 1780 CrossRef CAS;
(b) M. Liu, W. Liao, C. Hu, S. Du and H. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1585 CrossRef CAS PubMed;
(c) S. Du, C. Hu, J.-C. Xiao, H. Tan and W. Liao, Chem. Commun., 2012, 48, 9177 RSC;
(d) L. K. Batchelor, R. Shaw, S. J. Markey, M. Helliwell and E. J. L. McInnes, Chem.–Eur. J, 2010, 16, 5554 CrossRef CAS PubMed.
- For other examples see:
(a) C. E. Talbot-Eeckelaers, G. Rajaraman, J. Cano, G. Aromi, E. Ruiz and E. K. Brechin, Eur. J. Inorg. Chem., 2006, 3382 CrossRef CAS;
(b) M. Eshel and A. Bino, Inorg. Chim. Acta, 2002, 329, 45 CrossRef CAS.
- N. V. Gerbeleu, Y. T. Struchkov, G. A. Timco, A. S. Batsanov, K. M. Indrichan and G. A. Popovich, Dokl. Akad. Nauk SSSR, 1990, 313, 1459 CAS.
-
(a) M. L. Baker, G. A. Timco, S. Piligkos, J. M. Mathieson, H. Mutka, F. Tuna, P. Kozlowski, M. Antkowiak, T. Guidi, T. Gupta, H. Rath, R. J. Woolfson, G. Kamieniarz, R. G. Pritchard, H. Weihe, L. Cronin, G. Rajarman, D. Collison, E. J. L. McInnes and R. E. P. Winpenny, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(47), 19113 CrossRef CAS PubMed;
(b) D. M. Low, G. Rajaraman, M. Helliwell, G. Timco, J. van Slageren, R. Sessoli, S. T. Ochsenbein, R. Bircher, C. Dobe, O. Waldmann, H.-U. Güdel, M. A. Adams, E. Ruiz, S. Alvarez and E. J. L. McInnes, Chem.–Eur. J, 2006, 12, 1385 CrossRef CAS PubMed.
-
(a) R. A. Coxall, A. Parkin, S. Parsons, A. A. Smith, G. A. Timco and R. E. P. Winpenny, J. Solid State Chem., 2001, 159, 321 CrossRef CAS;
(b) D. Collison, M. Murrie, V. S. Oganesyan, S. Piligkos, N. R. J. Poolton, G. Rajaraman, G. M. Smith, A. J. Thomson, G. A. Timco, W. Wernsdorfer, R. E. P. Winpenny and E. J. L. McInnes, Inorg. Chem., 2003, 42, 5293 CrossRef CAS PubMed;
(c) E.
J. L. McInnes, S. Piligkos, G. A. Timco and R. E. P. Winpenny, Coord. Chem. Rev., 2005, 249, 2577 CrossRef CAS.
- F. A. Larsen, E. J. L. McInnes, H. El Mkami, J. Overgaard, S. Piligkos, G. Rajaraman, E. Rentschler, A. A. Smith, G. M. Smith, V. Boote, M. Jennings, G. A. Timco and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 101 CrossRef CAS.
-
(a) S. A. Oschsenbein, F. Tuna, M. Rancan, R. S. G. Davies, C. A. Muryn, O. Waldmann, R. Bircher, A. Sieber, G. Carver, H. Mutka, F. Fernandez-Alonso, A. Podlesnyak, L. P. Englehardt, G. A. Timco, H. U. Güdel and R. E. P. Winpenny, Chem.–Eur. J., 2008, 14, 5144 CrossRef PubMed;
(b) L. P. Englehardt, C. A. Muryn, R. G. Pritchard, G. A. Timco, F. Tuna and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2008, 47, 924 CrossRef PubMed;
(c) F. A. Larsen, J. Overgaard, S. Parsons, E. Rentschler, A. A. Smith, G. A. Timco and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 5978 CrossRef CAS PubMed.
-
(a) G. A. Timco, S. Carretta, F. Troiani, F. Tuna, R. J. Pritchard, C. A. Muryn, E. J. L. McInnes, A. Ghirri, A. Candini, P. Santini, G. Amoretti, M. Affronte and R. E. P. Winpenny, Nat. Nanotechnol., 2009, 4, 173 CrossRef CAS PubMed;
(b) G. A. Timco, T. B. Faust, F. Tuna and R. E. P. Winpenny, Chem. Soc. Rev., 2011, 40, 3067 RSC.
- C.-F. Lee, D. A. Leigh, R. G. Pritchard, D. Schultz, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Nature, 2009, 458, 314 CrossRef CAS PubMed.
-
(a) G. F. S. Whitehead, J. Fernando-Soria, L. G. Christie, N. F. Chilton, G. A. Timco, F. Moro and R. E. P. Winpenny, Chem. Sci., 2014, 5, 235 RSC;
(b) G. F. S. Whitehead, F. Moro, G. A. Timco, W. Wernsdorfer, S. J. Teat and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2013, 52, 9932 CrossRef CAS PubMed.
-
(a) R. Inglis, C. Milios, L. F. Jones, S. Piligkos and E. K. Brechin, Chem. Commun., 2011, 48, 181 RSC;
(b) R. Inglis, L. F. Jones, C. J. Milios, S. Datta, A. Collins, S. Parsons, W. Wernsdorfer, S. Hill, S. P. Perlepes, S. Piligkos and E. K. Brechin, Dalton Trans., 2009, 3403 RSC;
(c) C. J. Milios, R. Inglis, A. Vinslava, R. Bagai, W. Wernsdorfer, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 12505 CrossRef CAS PubMed.
-
(a) T. C. Stamatatos, K. V. Pringouri, C. P. Raptopoulou, R. Vicente, V. Psycharis, A. Escuer and S. P. Perlepes, Inorg. Chem. Commun., 2006, 9, 1178 CrossRef CAS;
(b) K. V. Pringouri, C. P. Raptopoulou, A. Escuer and T. C. Stamatatos, Inorg. Chim. Acta, 2007, 360, 69 CrossRef CAS;
(c) A.-R. Tomsa, Y. Li, S. Blanchard, P. Herson, K. Boubekeur, P. Gouzerh and A. Proust, J. Cluster Sci., 2014, 25, 825 CrossRef CAS;
(d) P. Chaudhuri, M. Hess, E. Rentschler, T. Weyhermüller and U. Flörke, New J. Chem., 1998, 22, 553 RSC.
-
(a) D. I. Alaxendropoulos, A. M. Mowson, M. Pilkington, V. Bekiari, G. Christou and T. C. Stamatatos, Dalton Trans., 2014, 43, 1965–1969 RSC;
(b) T. C. Stamatatos, D. Foguet-Albiol, C. C. Stoumpos, C. P. Raptopoulou, A. Terzis, W. Wernsdorfer, S. P. Perlepes and G. C. Christou, J. Am. Chem. Soc., 2005, 127, 15380 CrossRef CAS PubMed.
-
(a) B. N. Figgis and G. B. Robertson, Nature, 1965, 205, 694 CrossRef CAS;
(b) K. J. Schenk and H. S. Güdel, Inorg. Chem., 1982, 21, 2253 CrossRef CAS;
(c) F. A. Cotton and W. Wang, Inorg. Chem., 1982, 21, 2675 CrossRef CAS;
(d) M. K. Nagi, A. Harton, S. Donald, Y.-S. Lee, M. Sabat, C. J. O'Connor and J. B. Vincent, Inorg. Chem., 1995, 34, 3813 CrossRef CAS;
(e) R. D. Cannon, U. A. Jayasooriya, F. E. Sowrey, C. Tilford, A. Little, J. P. Bourke, R. D. Rogers, J. B. Vincent and G. J. Kearley, Inorg. Chem., 1998, 37, 5675 CrossRef CAS PubMed;
(f) R. D. Cannon and R. P. White, Prog. Inorg. Chem., 1998, 36, 195 CrossRef;
(g) V. Psycharis, C. P. Raptopoulou, A. K. Boudalis, Y. Sanakis, M. Fardis, G. Diamantopoulos and G. Papavassiliou, Eur. J. Inorg. Chem., 2006, 3710 CrossRef CAS;
(h) V. Corradini, C. Cervetti, A. Ghirri, R. Biagi, U. Del Pennino, G. A. Timco, R. E. P. Winpenny and M. Affronte, New J. Chem., 2011, 35, 1683 RSC.
- A. W. Addison, T. N. Rao, J. Reedjik, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans, 1984, 1349 RSC.
- For examples of extended networks comprising {Cr3O(carboxylate)x} nodes see:
(a) S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284 RSC;
(b) C. Serre, C. Mellot-Draznieks, S. Surblé, N. Auldebrand, N. Filinchuk and G. Férey, Science, 2007, 315, 1828 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- P. McArdle, P. Daly and D. Cunningham, J. Appl. Crystallogr., 2002, 35, 378 CrossRef CAS.
-
(a) A. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS;
(b) P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- R. Dunsten and T. A. Henry, J. Chem. Soc., Trans., 1899, 75, 66 RSC.
- G. Chaboussant, R. Basler, H.-U. Gudel, S. T. Ochsenbein, A. Parkin, S. Parsons, G. Rajaraman, A. Sieber, A. A. Smith, G. A. Timco and R. E. P. Winpenny, Dalton Trans., 2004, 2758 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1481774–1481778. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra14811e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence e,
Alan. G. Ryderb,
Stergios Piligkosd,
Euan K. Brechinc and
Leigh F. Jones*ab
e,
Alan. G. Ryderb,
Stergios Piligkosd,
Euan K. Brechinc and
Leigh F. Jones*ab