
Theoretical evaluation of some benzotriazole and phospono derivatives as aluminum corrosion inhibitors: DFT and molecular dynamics simulation approaches
Abstract
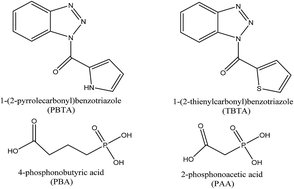
The adsorption and corrosion inhibition properties of some benzotriazole and phospono derivatives namely, 1-(2-pyrrolecarbonyl)-benzotriazole (PBTA), 1-(2-thienylcarbonyl)-benzotriazole (TBTA), 2-phosphonoacetic acid (PAA) and 4-phosphonobutyric acid (PBA) on the corrosion of aluminum were investigated by quantum chemical calculations and by molecular dynamics simulations. Global reactivity descriptors such as EHOMO, ELUMO, HOMO–LUMO energy gap (ΔE), chemical hardness (η), softness (σ), electronegativity (χ), proton affinity (PA), electrophilicity (ω) and nucleophilicity (ε) have been calculated and discussed. The analysis of the adsorption behavior of these mentioned inhibitors on the Al (111) surface was investigated using molecular dynamics simulations. The binding energies on the Al (111) surface of the studied compounds followed the order: PBTA > TBTA > PBA > PAA. The results obtained in this study are compatible with experimental data and it is proposed that the above mentioned molecules can be successfully used to prevent the corrosion of aluminum metal.
 Please wait while we load your content...
Please wait while we load your content...

