
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN,N-Bis-(dimethylfluorosilylmethyl)amides of N-organosulfonylproline and sarcosine: synthesis, structure, stereodynamic behaviour and in silico studies†
Alexey A.
Nikolin
*a,
Eugenia P.
Kramarova
a,
Alexander G.
Shipov
a,
Yuri I.
Baukov
a,
Vadim V.
Negrebetsky
a,
Dmitry E.
Arkhipov
b,
Alexander A.
Korlyukov
b,
Alexey A.
Lagunin
cd,
Sergey Yu.
Bylikin
e,
Alan R.
Bassindale
e and
Peter G.
Taylor
e
aDepartment of Chemistry, N.I. Pirogov Russian National Research Medical University, Ostrovityanov St. 1, Moscow 117997, Russian Federation. E-mail: nikson1111@yahoo.com
bA. N. Nesmeyanov's Institute of Organoelement Compounds, RAS, Vavilova St. 28, 119991 Moscow, Russian Federation
cDepartment of Bioinformatics, N.I. Pirogov Russian National Research Medical University, Ostrovityanov St. 1, Moscow 117997, Russian Federation
dInstitute of Biomedical Chemistry, Pogodinskaya Str., 10/8, Moscow, 119121, Russian Federation
eDepartment of Chemistry, Open University, Walton Hall, Milton Keynes, MK7 6AA, UK
First published on 25th July 2016
Abstract
(O→Si)-Chelate difluorides R3R2NCH(R1)C(O)N(CH2SiMe2F)2 (9a–c, R1R2 = (CH2)3, R3 = Ms (a), Ts (b); R1 = H, R2 = Me, R3 = Ms (c)), containing one penta- and one tetracoordinate silicon atoms were synthesized by silylmethylation of amides R3R2NCH(R1)C(O)NH2, subsequent hydrolysis of unstable intermediates R3R2NCH(R1)C(O)N(CH2SiMe2Cl)2 (7a–c) into 4-acyl-2,6-disilamorpholines R3R2NCH(R1)C(O)N(CH2SiMe2O)2 (8a–c) and the reaction of the latter compounds with BF3·Et2O. The structures of disilamorpholines 8a,c and difluoride 9a were confirmed by an X-ray diffraction study. According to the IR and NMR data, the O→Si coordination in solutions of these compounds was weaker than that in the solid state due to effective solvation of the Si–F bond. A permutational isomerisation involving an exchange of equatorial Me groups at the pentacoordinate Si atom in complexes 9a–c was detected, and its activational parameters were determined by 1H DNMR. In silico estimation of possible pharmacological effects and acute rat toxicity by PASS Online and GUSAR Online services showed a potential for their further pharmacological study.
Introduction
Hypercoordinate silicon compounds are the focus of intense research due to the diversity of their structures, chemical properties,1 stereodynamic behaviour2 and practical use in stereoselective synthesis3 and medical diagnostics.4 In recent years a large number of new types of pentacoordinate silicon compounds have been synthesized, including complexes with five different atoms in the silicon environment, compounds with SiO5, SiS2N2C, SiS2O2C, SiN4X (X = S, Se, Te) skeletons and others.5At the same time, certain classes of organosilanes containing both penta- and tetracoordinate silicon atoms in the same molecule remain virtually unknown. Among these compounds are N,N-bis(dimethylhalogenosilylmethyl)amides, where two silicon centres compete for a single carbonyl group. One of the Si atoms in these amides extends its coordination number to five and forms an (O→Si)-chelate ring while another Si atom remains tetracoordinate. Up to date, very few examples of such compounds have been reported,6 with the structures of only four complexes (1,6b2,6b3 (ref. 6e) and 4 (ref. 6e)) determined by X-ray method.
Since each of the two silicon atoms in dihalides 1–4 can potentially form a coordination bond with the carbonyl group, these compounds are particularly interesting as models for studying stereodynamic processes in solutions (such as alternating coordination or permutational isomerisation), pathways of SN2–Si reactions, relative contributions of the silicon centres to O→Si coordination and the effects of such coordination on the reactivity of SiIVMe2Hal and SiVMe2Hal groups within a single molecule.

Earlier we described (O→Si)-monochelate fluorosilanes RSO2-Pro-N(Me)CH2SiMe2F (5), containing an electron-withdrawing organosulfonyl group at the nitrogen atom of the amino acid fragment.7 In the present work, we report the synthesis, structures and stereodynamic behaviour of dinuclear fluorosilyl derivatives of proline and sarcosine R3R2NCH(R1)C(O)N(CH2SiMe2F)2 (9), synthesised by bis-silylmethylation of N-organosulfonyl-(S)-proline and N-mesylsarcosine amides R3R2NCH(R1)C(O)NH2 (6) via unstable dichlorides R3R2NCH(R1)C(O)N(CH2SiMe2Cl)2 (7) and isolable N-substituted 2,6-disilamorpholines R3R2NCH(R1)C(O)N(CH2SiMe2O)2 (8).
Discussion of the results
Synthesis of disilamorpholines
Disilamorpholines 8 were prepared by the general synthetic approach developed by us for various silacyclanes.6d,e,8 The starting compounds, primary amides 6, were silylmethylated by a mixture of chloro(chloromethyl)dimethylsilane and hexamethyldisilazane with subsequent hydrolysis of unstable dichlorides 7 into target 4-acyl-2,6-disilamorpholines 8 (Scheme 1).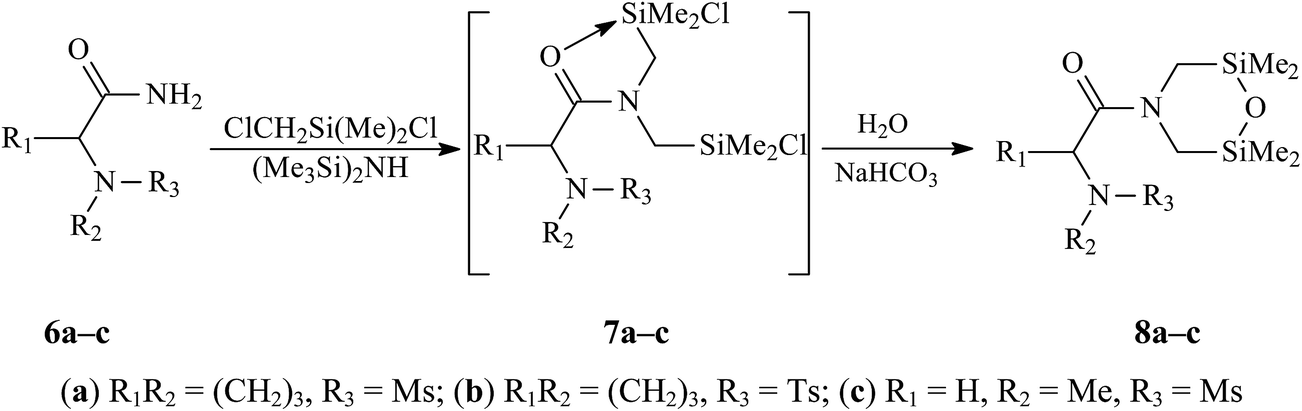 | ||
| Scheme 1 | ||
Mesyl and tosyl derivatives of (S)-proline, Ms-Pro-N(CH2SiMe2)2O (8a) and Ts-Pro-N(CH2SiMe2)2O (8b), and mesyl derivative of sarcosine, MsN(Me)CH2C(O)N(CH2SiMe2)2O (8c), were obtained by one-pot syntheses with yields of 75, 78 and 80%, respectively. The composition and structures of compounds 8 were confirmed by the elemental analysis, IR and multinuclear (1H, 13C, 29Si and CP/MAS 29Si) NMR spectroscopy. The structures of compounds 8a and 8c were also determined by X-ray method (see below).
The formation of hydrolytically unstable dichloride 7a was detected by IR spectroscopy. When a mixture of amide 6a with three equivalents of ClCH2SiMe2Cl and one equivalent of (Me3Si)2NH was refluxed in benzene or toluene, the absorption of the NCO fragment in 6a was gradually replaced by two absorptions (at 1590 and 1505 cm−1) of the same fragment in 7a, which was typical O→Si chelates of pentacoordinate silicon.6b,9 IR spectra of all 4-acyl-2,6-disilamorpholines 8a–c showed a strong absorption of the NCO fragment at 1630 cm−1.
In the 1H NMR spectra of chiral proline derivatives 8a,b, the signals of two SiMe2 groups appear as four singlets.
The 29Si NMR spectra of disilamorpholines 8a–c in solutions contain two signals at approximately 8 and 10 ppm, which are almost independent of the amino acid or N-substituent nature. The same chemical shifts of 29Si are observed in the solid-state CP/MAS spectra of these compounds (see Experimental section). Therefore, the solvation of tetracoordinate silicon atoms has no noticeable effect on their chemical shifts.
The above data suggest that both silicon atoms in compounds 8a–c are tetracoordinate.6c Similar to the double set of signals of SiMe2 groups in 1H NMR spectra, the presence of two signals in 29Si NMR spectra of these compounds is probably caused by the hindered amide rotation.
Synthesis of difluorides
In contrast to hydrolytically labile Si–Cl bonds in pentacoordinate dichlorides 7a–c, the Si–F bonds in their difluoro analogues 9a–c were expected to be more stable. (O→Si)-Chelate N′,N′-bis(dimethylfluorosilylmethyl)-N-organosulfonyl-(S)-prolinamides (9a,b) and N′,N′-bis(dimethylfluorosilylmethyl)-N-mesylsarcosinamide (9c) were prepared by the reaction of disilamorpholines 8a–c with BF3·Et2O in acetonitrile (Scheme 2). | ||
| Scheme 2 | ||
The composition and structure of difluorides 9a–c were determined by the elemental analysis, IR and multinuclear (1H, 13C and 29Si) NMR spectroscopy. The coordination states of both silicon atoms in compound 9a in the solid state was further confirmed by X-ray single-crystal study (see below) and 29Si CP/MAS NMR.
Multinuclear NMR spectroscopy
1H NMR spectra of difluorides 9a–c contain two signals of the SiMe2 groups in the upfield region. These signals can be attributed to specific SiMe2 groups using Bruker 2D pulse sequence {1H-29Si}HMBS. For example, the cross-peaks in the 2D spectrum of 9b (Fig. 1) indicate that the upfield signal of SiMe2 protons corresponds to the signal of pentacoordinate 29Si at −20 ppm while the downfield signal of SiMe2 protons corresponds to the signal of tetracoordinate 29Si at +30 ppm.2b,c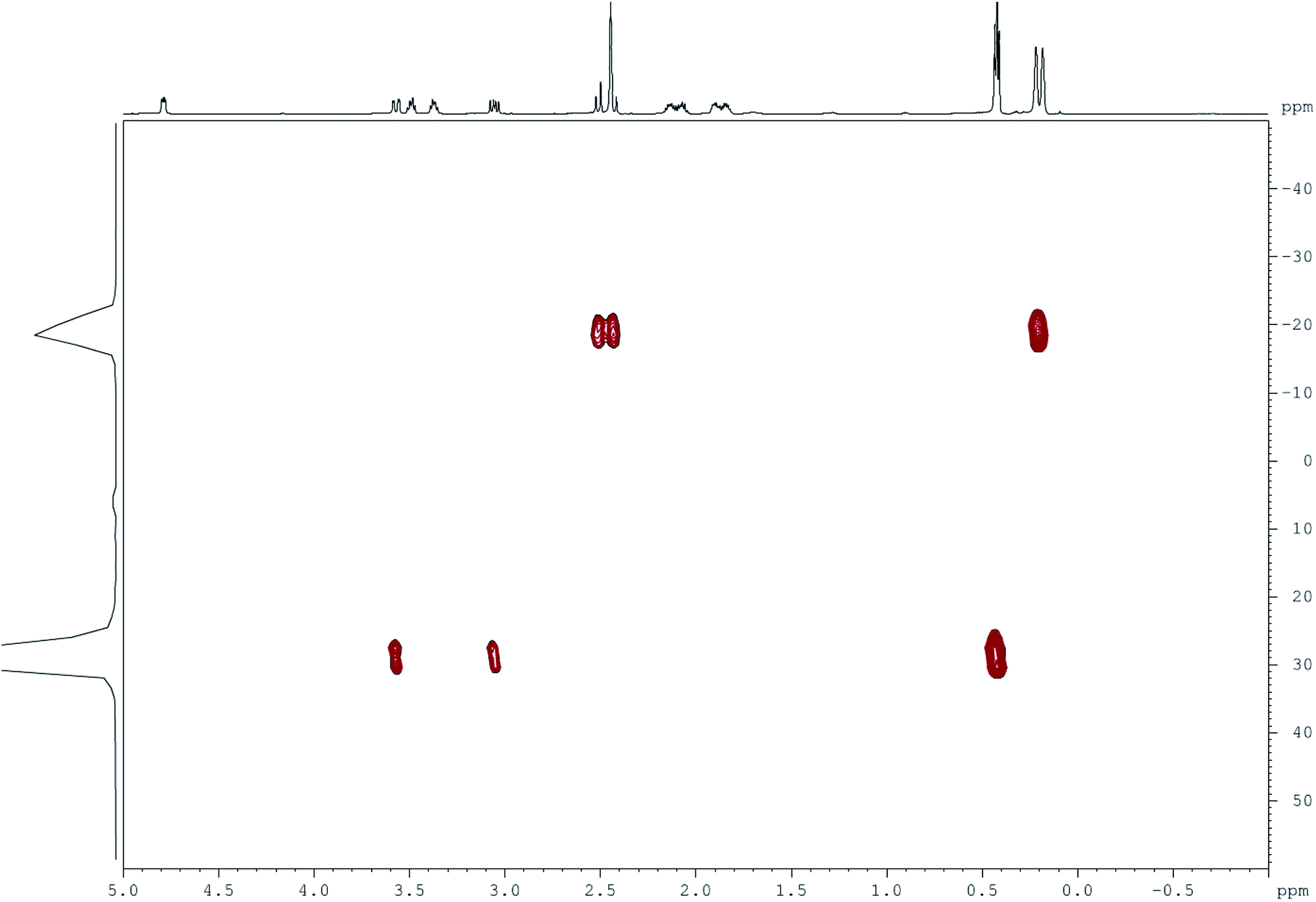 | ||
| Fig. 1 Two-dimensional NMR spectrum of 9b (Bruker {1H-29Si}HMBS, CDCl3, 600 MHz). | ||
Direct spin–spin coupling constants 1JSiF in NMR spectra of compounds 9 for tetracoordinate silicon (230–260 Hz) were generally lower than those for pentacoordinate silicon (ca. 280 Hz; see Experimental section). Such difference, observed both in solutions and solid state, reflected the weakening of the Si–F bond at SiV in comparison with SiIV (see X-ray data for 9a).2b,c For the same reason, the spin–spin coupling constant 3JHF was observed at ambient temperature only for the SiIVMe2 group but not for the SiVMe2 group. Finally, the weakening of the SiV–F bonds affected the 2JCF constants in 13C NMR spectra: the observed spin–spin coupling frequencies at SiV centres (10–15 Hz) were significantly lower than those at the SiIV centres (ca. 30 Hz).
Intramolecular O→Si coordination in complexes 9 in solutions was further confirmed by the down field shift of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O signal in their 13C NMR spectra. The characteristic patterns of SiV(CH3)2 and NCH2SiV signals in 1H NMR spectra of difluorides 9a,b (two singlets of equal intensity and an AB-system quartet, respectively) indicated the presence of a chiral carbon atom in their molecules.
O signal in their 13C NMR spectra. The characteristic patterns of SiV(CH3)2 and NCH2SiV signals in 1H NMR spectra of difluorides 9a,b (two singlets of equal intensity and an AB-system quartet, respectively) indicated the presence of a chiral carbon atom in their molecules.
The 29Si signals in solid-state NMR spectra of compounds 9 had greater upfield shifts (ca. −40 ppm) than the same signals in solutions (ca. −10 ppm). Similar effect was observed for monofluorides 5 and was probably caused by effective solvation of pentacoordinate silicon.7a
Using the difference between the observed chemical shifts of a SiV atom and the typical chemical shift of a SiIV atom (ca. 30 ppm), the coordination contribution (−Δδ = δSiV − δSiIV)2a in difluorides 9 can be estimated to be approximately 50 ppm. The comparison of this value to coordination contributions in chlorosilanes RSO2-Pro-N(Me)CH2SiMe2Cl (70–75 ppm), silyloxonium halides [R-Pro-N(Me)CH2SiMe2OH2]X (R = AlkSO2, ArSO2, Ac; X = Cl, Br) (70–80 ppm)10 and theoretical data for monofluorosilanes 5 (ref. 7b) and MeC(O)N(Me)CH2SiMe2F11 (same as above) indicates a relatively weak coordination in difluorides 9.
All difluorides have two signals in their 19F NMR spectra: one at approximately −159 ppm and another at −119 ÷ −125 ppm. According to literature data, these signals belong to SiIVMe2F and SiVMe2F groups, respectively.2b,c
Variable-temperature 1H, 19F and 29Si NMR studies
The strength of intramolecular coordination in monochelates of pentacoordinate silicon strongly depends on the nature of the substituent X (Scheme 3; see2c,7a and references therein). | ||
| Scheme 3 | ||
In the case of compounds with the OSiC3X coordination set and X = Hal or OTf, structures A and B are typical for fluorides, C for chlorides, D for bromides, and E for iodides and triflates.
To study the temperature effects on the coordination set structure in difluorides 9a–c, the temperature-dependent 1H, 19F and 29Si NMR spectra of these compounds in CDCl3 were obtained. The decrease in temperature from +20 to −60 °C led to reversible downfield shifts of 1H and 19F signals (by ca. 0.03 and 3–4 ppm, respectively) of the SiVMe2F group. At the same time, the chemical shift of 19F in the SiIVMe2F group was not affected by the temperature. Such behaviour of 1H and 19F signals suggests an increased contribution of form B (Scheme 3) at low temperatures.
Similar to monofluorosilanes,7a the increase in temperature to +60 °C caused very small reversible broadening of the SiMe2 and NCH2 signals in 1H NMR spectra of difluorides 9a–c. Such broadening was indicative of a permutational isomerisation at the SiV coordination set of these compounds.
The activation parameters of the permutation were calculated by a 1H DNMR method using a full line-shape analysis of the signals. For all studied compounds, the stereodynamic processes in CDCl3 were characterised by a narrow range of activation energies (∼24 kcal mol−1 or greater) and high negative values of the entropy of activation (ca. ∼ −20 cal mol−1 K−1). These values were very similar to the activation parameters of N-(dimethylfluorosilylmethyl)- and N-[fluoro(methyl)(phenyl)silylmethyl]amides and -lactams,2a,12–15 as well as RSO2-Pro-N(Me)CH2SiMe2F (5),6a where R = Me, Ph, 4-MeC6H4, 4-ClC6H4, 4-BrC6H4 or 4-NO2C6H4.
XRD studies
Disilamorpholine 8a (Fig. 2) crystallizes in two polymorph modifications (8a and 8a′). | ||
| Fig. 2 Molecular structure of 8a with thermal ellipsoids shown at the 50% probability level. | ||
The orthorhombic (P212121) crystals 8a were obtained from a heptane–benzene mixture with a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, whereas monoclinic (P21) crystals 8a′ were obtained from ethanol. There are two crystallographically independent molecules in the asymmetric unit of 8a; its volume is 3.84 times larger than that of 8a′, because the cell 8a′ contains a void of about 40 Å3. The structure of the 2,6-disilamorpholine fragment in compounds 8a, 8a′ and 8c (Fig. 3) is analogous to the previously published five structures (CSD refcodes:16 QOMTAN, QOMTER, XATQIT, XULNAT, XULNEX).
:1, whereas monoclinic (P21) crystals 8a′ were obtained from ethanol. There are two crystallographically independent molecules in the asymmetric unit of 8a; its volume is 3.84 times larger than that of 8a′, because the cell 8a′ contains a void of about 40 Å3. The structure of the 2,6-disilamorpholine fragment in compounds 8a, 8a′ and 8c (Fig. 3) is analogous to the previously published five structures (CSD refcodes:16 QOMTAN, QOMTER, XATQIT, XULNAT, XULNEX).
 | ||
| Fig. 3 Molecular structure of 8c with thermal ellipsoids shown at the 50% probability level. | ||
The mesyl group and 2,6-disilamorpholine fragment have syn-conformation relative to the proline ring: the corresponding torsion angles C8–N2–S1–C12 and C20–N4–S2–C24 in 8a are 95.3(2)° and 88.8(2)° for two crystallographically independent molecules, respectively, and the torsion angle C8–N2–S1–C12 in 8a′ is 94.7(5)°.
An asymmetric unit of difluoride 9a contains two crystallographically independent molecules, which differ by mutual orientation of the proline moiety and Me2FSiCH2 group relative to the chelate ring. In the case of syn-conformation, the interatomic distance S1⋯Si2 is 5.387(1) Å, whereas for anti-configuration the distance S2⋯Si4 is 6.340(1) Å. In 9a (Fig. 4), one of the silicon atoms is pentacoordinated, and its coordination polyhedron is a distorted trigonal bipyramid (axial angles O1–Si1–F1 and O4–Si3–F3 are 172.1(1)° and 171.9(1)°, the deviations of Si1 and Si3 atoms from the planes of equatorial substituents toward fluorine atoms are 0.167(1) Å and 0.176(1) Å for two crystallographically independent molecules, respectively).
 | ||
| Fig. 4 Molecular structure of 9a with thermal ellipsoids shown at the 50% probability level. | ||
The structures of coordination polyhedra of Si1 and Si2 atoms in 9a are noticeably different from those in the series of (O→Si)-chelate N′-(dimethylfluorosilylmethyl)-N′-methyl-N-(organosulfonyl)prolinamides,7a complexes 1 (ref. 6d) and 3 (ref. 6e) (selected bond lengths are given in Table 1).
The structure of 3 differs significantly from other difluorides due to the coordination of amide oxygen atom with the difluoroboron group while the coordination with the silicon atom is very weak. Thus, the axial Si–O bonds are shortened by 0.07–0.17 Å, and SiV–F bonds lengthened by 0.02–0.04 Å compared to similar bonds in prolinamide derivatives and difluoride 1. The SiIV–F bonds are lengthened by 0.05–0.10 Å in comparison with similar bonds in difluorides 1 and 3. Atoms Si2 and Si4 are not coordinated by any oxygen atoms, with the shortest intermolecular contact Si4⋯O2 of 3.538(1) Å.
Quantum-chemical studies of the permutational isomerization
To test the applicability of the mechanism (Scheme 4) previously suggested for the permutational isomerisation of N-(dimethylfluorosilylmethyl)amides1 to N,N-bis-(dimethylfluorosilylmethyl)amides, we carried out quantum chemical studies of molecule 9a.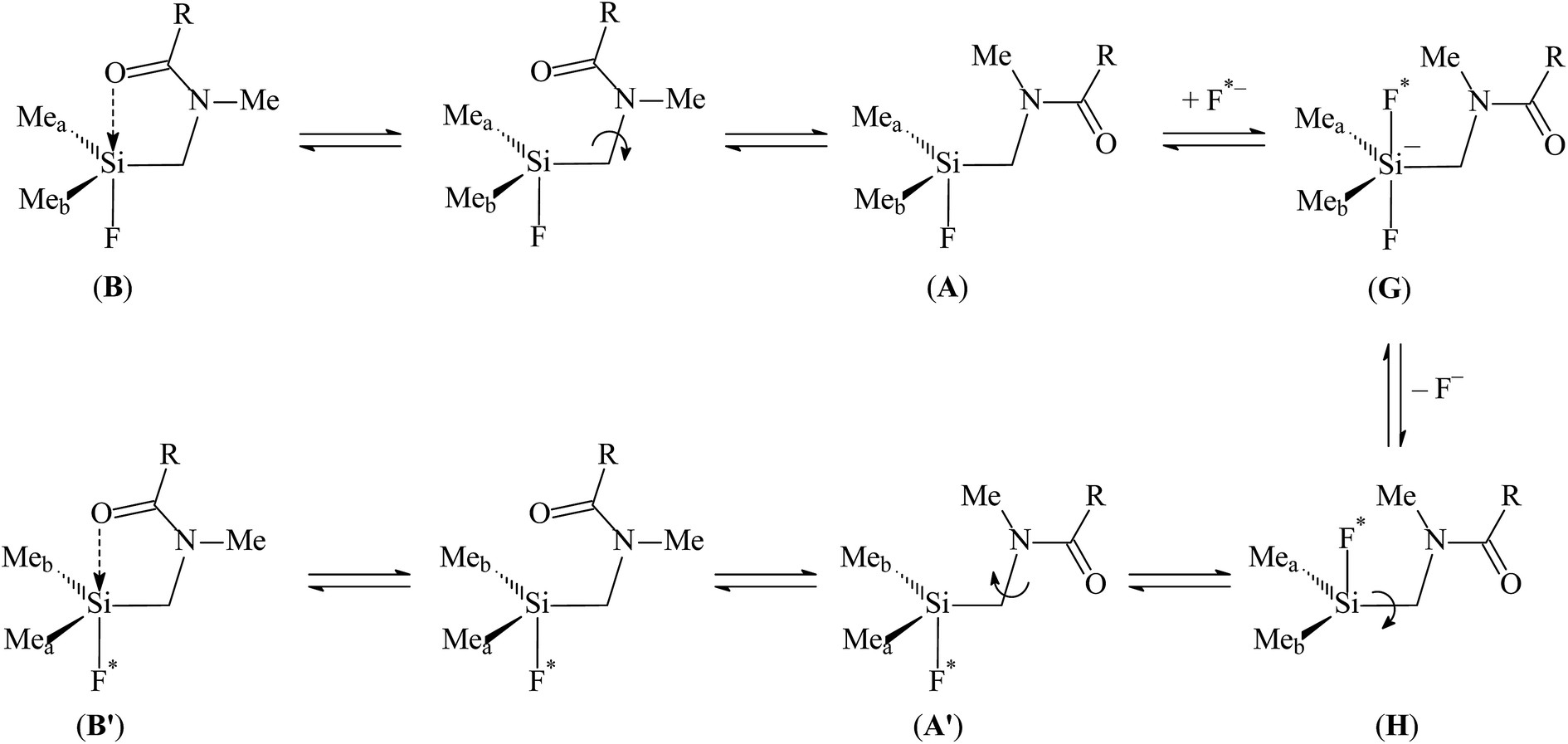 | ||
| Scheme 4 | ||
At higher temperatures, the equilibrium B ⇌ A (Schemes 3 and 4) shifts towards the tetracoordinate topomer A. The nucleophilic attack at the Si atom by a fluoride anion (F*−) produces pentacoordinate difluoride G, which subsequently loses the F− anion and forms tetracoordinate intermediate H. The rotation around the Si–CH2 bond produces topomer A′ and finally complex B′ with inverted orientation of the methyl groups at silicon.
According to our previous study, the external fluorine anion can attack tetracoordinated silicon, and the dissociation energy of resulting Si–F bond in gas phase is equal to ∼90 kcal mol−1. Solvation of the F− anion leads to significant decrease of the Si–F dissociation energy. It is reasonable to assume that similar processes can occur in solution of 9a in CDCl3. Due to the presence of two dimethylfluorosilylmethyl and one bulky tosyl groups, the silicon atoms seems to be less accessible for nucleophilic attack as compared to N-(dimethylfluorosilylmethyl)amides, where only one dimethylfluorosilylmethyl group is present. Hence, the stereodynamic processes in solution of 9a can be more complex as compared to N-(dimethylfluorosilylmethyl)amides.7a
An alternative mechanism can involve the carbonyl group migration from one dimethylfluorosilyl to another (similarly to derivatives urea17) (Scheme 5).
 | ||
| Scheme 5 | ||
In any case, the cleavage of the Si–O coordination bond and the certain conformational changes are necessary to the transfer the carbonyl oxygen atom from one dimethylfluorosilylmethyl to another. Thus, the detailed inspection of these processes can be very useful for understanding the nature of permutational isomerisation in the solution of 9a.
Quantum-chemical calculations of 9a were carried out using Gaussian 03W program.18 Hybrid PBE0 functional and 6-311G(d,p) basis set were utilized for structure optimization, hessian calculations, relaxed potential energy scans and transition state search. To account for the effect of nonspecific solvation, the PCM model was applied (the value of dielectric constant corresponded to chloroform). All calculations was performed with tight optimization criteria (Opt = tight) and precise grid for computation of two-electron integrals (Int(Grid = Ultrafine)). Molecular graphics was drawn with ChemCraft program.19 General views of calculated structures, atomic coordinates and total energies can be found in ESI.†
Analysis of potential energy surface for 9a in its isolated molecule and CDCl3 (PCM calculation) has shown that the presence of two conformational isomers correspond to the cyclic structures (where the Si–O coordination bond is present) and two other conformers belong are acyclic (Si–O coordination bond is absent). According to quantum chemical calculations, the influence of dielectric continuum used in PCM model leads to significant changes in molecular structure of 9a. The most noticeable change is the decrease of Si1⋯O1 distance from 2.36–2.37 to 2.25 Å. Cyclic conformers are more favourable as compared to the acyclic conformers. The difference between two isolated most stable cyclic and acyclic structures is 2.51 kcal mol−1. The use of PCM model for the description of solvation increases this difference to 4.37 kcal mol−1, which is in good agreement with our earlier calculations.7a All cyclic conformers can be characterized by the same geometry of coordination polyhedra of silicon atoms, so their 19F and 29Si chemical shifts should be very close.
Other differences are related to mutual orientation of N-organosulfonyl and dimethylfluorosilyl groups. In isolated cyclic and acyclic forms of 9a, these fragments are much closer to each other than in the solution. In two conformers (9a-cyclic2 and 9a-acyclic2, Fig. 5S and 6S, see ESI†), the Si2⋯O2 distances between one of the SiMe2F groups and the oxygen atom of the sulfonyl group are 3.698 and 3.720 Å, respectively. The optimization of these conformers in terms of PCM model (9a-cyclic2-CDCl3 and 9a-acyclic2-CDCl3, Fig. 7S and 8S†) increases the separation of the above fragments (the Si2⋯O2 distances become 4.533 and 3.962 Å). Conformers 9a-cyclic and 9a-acyclic (Fig. 1S and 2S†) are stabilized by weak C–H⋯O bonds between sulfonyl and methyl groups, so the Si2⋯O2 distances are 3.724 and 4.450 Å. Again, the application of PCM model increases Si2⋯O2 distances to 4.292 and 4.376 Å (9a-cyclic-CDCl3 and 9a-acyclic-CDCl3, Fig. 3S and 4S†). Thus, the effect of nonspecific solvation prevents the formation of Si2⋯O2 interactions, so the permutational isomerisation involving the sulfonyl group is unlikely to take place.
The information about the barrier of rotation around Si1–C3 and N1–C4 bonds can be useful to understand the mechanism of permutational isomerisation of 9a. These barriers were calculated by the relaxed potential energy surface scan of CNCO and F1Si1C3N1 torsion angles (the plots of the energy vs. scan coordinate are placed in ESI (Fig. 9S and 10S†)). The value of the rotation barrier around the Si1–C3 bond in isolated molecule 9a is approximately 7 kcal mol−1 (Fig. 9S†), so the rotation around the Si–C bond is possible despite the presence of an Si1–O1 coordination bond. In solution, the value of this barrier is even lower than that in isolated molecule (∼5.1 kcal mol−1). It is not surprising that the rotation around the N1–C4 bond is less favourable than the rotation around the Si1–C3 bond. Firstly, the N1–C3 bond is intermediate between ordinary and double (Table 2). Secondly, the rotation around the N1–C3 bond is attributed to the formation and cleavage of Si1–O1 and Si2–O1 coordination bonds. Our calculation gave the values of 26.3 kcal mol−1 for isolated molecule 9a and 24.8 kcal mol−1 for its solution in chloroform (Fig. 10S†). These values are very close to the permutational barriers measured for 9a–c by 19F DNMR study. Thus, the internal rotation can be responsible for the permutational isomerisation of 9a. Additional justification for this assumption was obtained by the localization of transition states (Fig. 11S and 12S†). The modes of negative vibrations (−67.0 and −62.8 cm−1 for isolated molecule and CDCl3 solution, respectively) correspond to the rotation around the N1–C4 bond and formation/dissociation of Si–O coordination bonds. The difference between energies of the most favourable cyclic conformers and transition state is 28.4 and 29.3 kcal mol−1 for isolated molecules and solution of 9a, respectively. These values are in agreement with the results of DNMR study. At the same time, the ΔS value calculated as the difference between the transition state and cyclic isomer is ∼−2 kcal mol−1 K−1, which is much lower than the experimental value. In our opinion, this difference can be explained by specific solvation (for instance, H-bonds between CDCl3 and carbonyl or sulfonyl groups, which can be responsible for stabilization of particular conformers).
| Conformer | Si1⋯O1 | Si2⋯O2 | Si1–F1 | Si2–F2 | O1Si1F1 |
|---|---|---|---|---|---|
| 9a-cyclic | 2.368 | 4.450 | 1.665 | 1.632 | 169.20 |
| 9a-acyclic | 3.130 | 3.724 | 1.638 | 1.646 | 79.69 |
| 9a-cyclic-CDCl3 | 2.248 | 4.533 | 1.686 | 1.640 | 170.25 |
| 9a-acyclic-CDCl3 | 3.222 | 3.962 | 1.652 | 1.644 | 78.82 |
| 9a-cyclic2 | 2.356 | 3.698 | 1.667 | 1.637 | 168.96 |
| 9a-acyclic2 | 2.984 | 3.720 | 1.637 | 1.639 | 76.09 |
| 9a-cyclic2-CDCl3 | 2.250 | 4.292 | 1.686 | 1.643 | 169.97 |
| 9a-acyclic2-CDCl3 | 3.175 | 4.376 | 1.644 | 1.644 | 77.90 |
| 9a-ts | 4.828 | 3.969 | 1.638 | 1.637 | 112.44 |
| 9a-ts-CDCl3 | 4.947 | 3.777 | 1.645 | 1.648 | 115.07 |
In silico estimation of possible pharmacological applications
Possible applications of synthesized complexes were evaluated by the search for similar compounds with known activities and computational prediction of biological activity based on “structure–activity” relationships (SAR) models. Such analysis provides a reasonable basis for planning further experimental studies of biological activity.In this study, we used the PubChem structural search for identification of equivalent and similar structures (https://pubchem.ncbi.nlm.nih.gov).20 The similarity was assessed by the Tanimoto equation and the PubChem dictionary-based binary fingerprint analysis (https://pubchem.ncbi.nlm.nih.gov/search/help_search.html). The search results for similar compounds are shown in Table 3.
| ID | Hits (probability) | The most similar compound with data on patents or activity |
|---|---|---|
| a Hits – number of similar compounds (≥90% or ≥ 80% Tanimoto index); CID – PubChem Compound ID. | ||
| 8a | 0 (90%); 161 (80%) | N-[(3S)-1-Methyl-2-oxopiperidin-3-yl]-N-(2-oxopropyl)methanesulfonamide (CID 58869395) |
| Patent description: sulfonylaminovalerolactams and derivatives thereof as factor Xa inhibitors | ||
| 8b | 216 (90%) | 4,4′-Ethylenebis(1-methyl-2,6-piperazinedione) (CID 97592) |
| Patent description: novel drug targets to overcome de novo drug-resistance in multiple myeloma; method of reducing amyloid-beta peptide levels using a bisdioxopiperazine; abatement process for contaminants; bis-dioxopiperazines and their use as protection agents bis-dioxopiperazines and their use as protection agents | ||
| Known activity: small molecule inhibitors of FGF22-mediated excitatory synaptogenesis & epilepsy measured in biochemical system using RT-PCR – 7012-01_Inhibitor_SinglePoint_HTS_Activity | ||
| 8c | 0 (90%); 19 (80%) | 2-(4-Acetylpiperazin-1-yl)-N-methylsulfonylacetamide (CID 89504348) |
| Patent description: dual-acting antihypertensive agents having angiotensin II type receptor antagonist activity and neprilysin-inhibition activity | ||
| 9a | 0 (90%); 74 (80%) | (1) N-(4-Amino-5-oxo-5-pyrrolidin-1-ylpentyl)methanesulfonamide (CID 17960593) |
| Patent description: alpha-amino acid sulphonyl compounds | ||
| (2) (2S)-1-[2-[Methyl(methylsulfonyl)amino]ethyl]pyrrolidine-2-carboxamide (CID 57572120) | ||
| Patent description: quinolinone compounds as 5-HT4 receptor agonists | ||
| 9b | 59 (90%) | Azepan-1-yl-[1-(4-methylphenyl)sulfonylpyrrolidin-2-yl]methanone (CID 2964486) |
| Known activity: active in HTS assay for activators of cytochrome P450 2A9 | ||
| 9c | 0 (90%); 10 (80%) | N,N-Dimethyl-2-[methyl(methylsulfonyl)amino]acetamide (CID 57682568) |
| Patent description: HIV integrase inhibitors | ||
According to Table 3, the studied complexes have different similar compounds with variable known activities. No two complexes have the same most similar compound, which could indicate their similar biological potentials.
Computational prediction of biological activity for studied complexes was carried out using SAR-based online services. Possible therapeutic effects and mechanisms of action were evaluated by PASS Online21 (http://www.way2drug.com/PASSOnline) while the LD50 values for acute rat toxicity were estimated by GUSAR Online22 (http://www.way2drug.com/gusar/acutoxpredict.html). The results of these predictions are summarized in Table 4.
| ID | Top 5 predicted therapeutic effects with probability > 50% | Top 5 predicted mechanisms of action with probability > 50% | Predicted LD50 values in mg kg−1, type of administration, class of toxicity |
|---|---|---|---|
| a IP – intraperitoneal route of administration; IV – intravenous route of administration; PO – oral route of administration; SC – subcutaneous route of administration; out of AD – compound is out of applicability domain of QSAR models. | |||
| 8a | Not predicted | Acetylcholine neuromuscular blocking agent | IP – out of AD, 127, IV, 4 class, 904, PO, 4 class, SC – out of AD |
| 8b | Not predicted | Acetylcholine neuromuscular blocking agent | IP – out of AD, 96, IV, 4 class, 1270, PO, 4 class, SC – out of AD |
| 8c | Spasmolytic | Acetylcholine neuromuscular blocking agent, anaphylatoxin receptor antagonist | IP – out of AD, 156, IV, 4 class, 453, PO, 4 class, 250, SC, 4 class |
| 9a | Antianginal, multiple sclerosis treatment, antiparkinsonian, neurodegenerative diseases treatment | Phosphatidylinositol 3-kinase C2beta inhibitor, RANTES antagonist, insulin growth factor agonist, insulin like growth factor 1 agonist | IP – out of AD, 80, IV, 4 class, PO – out of AD, 487, SC, 4 class |
| 9b | Antianginal, cardiovascular analeptic, multiple sclerosis treatment, cell adhesion molecule inhibitor | Integrin alpha2 antagonist, calmodulin antagonist, nicotinic alpha4beta4 receptor agonist | IP – out of AD, 101, IV, 4 class, PO – out of AD, SC – out of AD |
| 9c | Antianginal | Anaphylatoxin receptor antagonist, phospholipid-translocating ATPase inhibitor, 2-haloacid dehalogenase inhibitor, glycosylphosphatidylinositol phospholipase D inhibitor, NADPH peroxidase inhibitor | IP – out of AD, 130, IV, 4 class, PO – out of AD, 225, SC, 4 class |
The prediction results suggest that synthesized compounds may possess cardiovascular and CNS properties. Low levels of predicted acute rat toxicity makes them suitable for all routes of administration.
Conclusions
New difluorides R3R2NCH(R1)C(O)N(CH2SiMe2F)2 (9a–c) with one pentacoordinate and one tetracoordinate silicon atoms were synthesized by silylmethylation of amides R3R2NCH(R1)C(O)NH2, subsequent hydrolysis of unstable intermediates R3R2NCH(R1)C(O)N(CH2SiMe2Cl)2 (7a–c) into 4-acyl-2,6-disilamorpholines R3R2NCH(R1)C(O)N(CH2SiMe2O)2 (8a–c) and the reaction of the latter compounds with BF3·Et2O. According to IR and NMR data, the O→Si coordination in solutions of these compounds was weaker than in the solid state due to effective solvation of the Si–F bond. The absence of spin–spin coupling constants 3JHF of the methyl groups at SiV and their retention at SiIV indicates a significant weakening of the Si–F bond at pentacoordinate silicon, which favours its ionization. Based on in silica analysis, the synthesized compounds show a potential for pharmacological studies.Experimental section
IR-spectra of compounds in solution and in the solid state were recorded on a Bruker Tensor-27 spectrometer using KBr cells and an APR element, respectively. 1H, 13C and 19F NMR spectra in CDCl3 and DMSO-d6 were recorded on a Bruker Avance II 300 (1H, 300 MHz; 13C, 75.6 MHz; 19F, 282.2 MHz) and Jeol JNM-EX400 (1H, 400 MHz; 13C, 100.6 MHz; 19F, 376.3 MHz) instruments using standard pulse sequences. 29Si NMR spectra were recorded using the 1H-29Si HSQC pulse sequence supplied with the Bruker Avance II 600 instrument.23 The 1H, 13C, 29Si chemical shifts were measured using Me4Si as internal reference. The 19F chemical shifts were measured using BF3 as external reference. Negative values are to high field. 29Si NMR CP/MAS spectra in the solid state were recorded on a Jeol JNM-EX-400 instrument using 5 mm zirconia rotors and a Doty probe.The temperature calibration of the NMR spectrometers was performed by measuring the differences in chemical shifts between non-equivalent protons in methanol (−90…+30 °C) and ethyleneglycol (+30…+85 °C).24 The activational parameters of the permutational isomerisation were calculated using DNMR-SIM software25 and a modified Eyring equation.26 In each case, at least twelve temperature points were obtained to achieve a correlation coefficient of 0.997–0.999.
Chloro(chloromethyl)dimethylsilane, (S)-proline hydrochloride, sarcosine and all solvents were purchased from Acros and Sigma-Aldrich. Ethyl esters of N-mesyl-(S)-proline and N-tosyl-(S)-proline were synthesised as described earlier.10
Ethyl-N-mesyl-N-methylglycinate
Thionyl chloride (83.3 g, 0.27 mol) was added dropwise to a solution of N-methylglycine (44.5 g, 0.50 mol) in absolute ethanol (200 mL). The mixture was refluxed for 5 h, then the volatiles were removed in vacuum. The residue was suspended in an ice-cold mixture of water (20 mL) and diethyl ether (100 mL), and a solution of potassium hydroxide (28.0 g, 0.50 mol) in water (20 mL) was added over 5 min at 0 °C, followed by 250 g of anhydrous potassium carbonate. The organic layer was separated, the residue was washed with ether (2 × 50 mL), and the combined organic solutions were dried over magnesium sulfate. The solvent was removed in vacuum, and the residue was distilled to afford 38.0 g (65%) of ethyl N-methylglycinate with b.p. 43–45 °C (12 torr) and nD (ref. 20) 1.4105. Literature data:27 b.p. 46 °C (12 torr), nD (ref. 20) 1.4144.Methanesulfonyl chloride (11.5 g, 0.10 mmol) was added dropwise to a cooled solution of ethyl-N-methylglycinate (11.7 g, 0.10 mol) and triethylamine (10.1 g, 0.10 mol) in diethyl ether (80 mL). The mixture was stirred at ambient temperature for 2 h, the precipitate formed was filtered off, washed with ether (15 mL), and the combined organic solutions were evaporated in vacuum. The residue was distilled to afford 13.7 g (70%) of ethyl-N-mesyl-N-methylglycinate with b.p. 144–145 °C (9 torr) and m.p. 34–35 °C. IR spectrum (KBr, ν, cm−1): 1750 (CO), 1360 and 1160 (SO2). 1H NMR spectrum (CDCl3, δ, ppm (J, Hz)): 1.25 (3H, t, 3J 7.3, CH2CH3); 2.77 (3H, s, CH3N); 2.87 (3H, s, CH3S); 4.05 (2H, s, NCH2). 13C NMR spectrum (CDCl3, δ, ppm): 8.9 (CH2CH3); 35.3 (CH3N); 38.1 (CH3S); 51.4 (NCC(O)); 55.5 (CH2CH3); 173.9 (CO). Found, %: C 37.08; H 6.65; N 7.11. C6H13NO4S. Calculated, %: C 36.91; H 6.71; N 7.17.
N-Mesyl-(S)-prolinamide (6a)
Ethyl ester of N′-mesyl-(S)-proline (6.6 g, 30 mmol) was stirred with 50 mL of a 25% aqueous ammonia solution for 5 days at ambient temperature. The precipitate formed was isolated by filtration, dried in the open air and used without further purification. Yield 5.5 g (96%), m.p. 156–157 °C (from EtOH), [α]25D −101.3° (c 1.93, H2O). IR spectrum (KBr, ν, cm−1): 3449, 3170 (NH2); 1619 (NCO), 1321 and 1140 (SO2). 1H NMR spectrum (DMSO-d6, δ, ppm (J, Hz)): 1.75–2.25 (4H, m, 3,4-CH2); 2.83 (3H, s, CH3); 3.25–3.47 (2H, m, 5-CH2); 3.96–4.09 (1H, m, 2-CH); 6.1 and 6.7 (2H, two broad s, NH2). 13C NMR spectrum (DMSO-d6, δ, ppm): 22.0 (Me); 26.0 (C-4); 32.3 (C-3); 50.7 (C-5); 63.5 (C-2); 175.8 (CO). Found, %: C 37.35; H 6.39; N 14.50. C6H12N2O3S. Calculated, %: C 37.49; H 6.29; N 14.57.
N-Tosyl-(S)-prolinamide (6b)
Prepared similar to 6a. Yield 6.2 g (93%), m.p. 161–162 °C (from EtOH), [α]25D −134.6° (c 1.06, H2O). IR spectrum (KBr, ν, cm−1): 1643 (NCO), 1344, 1156 (SO2). 1H NMR spectrum (DMSO-d6, δ, ppm (J, Hz)): 1.31–1.83 (4H, m, 3,4-CH2); 2.43 (3H, s, CH3); 3.11–3.22 and 3.35–3.55 (2H, m, 5-CH2); 3.91–4.01 (1H, m, 2-CH); 5.95 and 6.71 (2H, two broad s, NH2); 7.36 (2H, d, 3J = 8.3, H Ar); 7.72 (2H, d, 3J 8.3, H Ar). 13C NMR spectrum (DMSO-d6, δ, ppm): 22.0 (Me); 25.7 (C-4); 31.8 (C-3); 50.9 (C-5); 63.7 (C-2); 129.2 (C-3,5 Ar); 131.4 (C-2,6 Ar); 135.4 (C-1 Ar); 145.8 (C-4 Ar); 175.5 (CO). Found, %: C 37.58, H 6.27, N 14.50. C6H12N2O3S. Calculated, %: C 37.49, H 6.29, N 14.57.
N-Mesyl-N-methylglycinamide (6c)
Prepared similar to 6a. Yield 4.1 g (82%), m. p. 170–171 °C (EtOH). IR spectrum (KBr, ν, cm−1): 3315, 3170 (NH2); 1657 (NCO), 1320 and 1150 (SO2). 1H NMR spectrum (DMSO-d6, δ, ppm (J, Hz)): 2.79 (3H, s, CH3); 2.87 (3H, s, CH3S); 3.58 and 3.71 (2H, two s, NCH2); 5.5 and 6.1 (2H, two broad s, NH2). 13C NMR spectrum (DMSO-d6, δ, ppm): 34.3 (CH3N); 37.1 (CH3S); 50.9 (NCC(O)); 174.9 (CO). Found, %: C 29.18, H 5.92, N 16.81. C4H10N2O3S. Calculated, %: C 28.91, H 6.06, N 16.86.
2,2,6,6-Tetramethyl-4-[N-mesyl-(S)-prolinyl]-2,6-disilamorpholine (8a)
A mixture of 6a (0.96 g, 5 mmol), hexamethyldisilazane (0.81 g, 5 mmol), chloro(chloromethyl)dimethylsilane (2.15 g, 15 mmol) and toluene (10 mL) was refluxed for 4 h, then allowed to cool down, and the precipitate formed was filtered out. The remaining solution was evaporated in vacuum, the residue was dissolved in chloroform (30 mL) and stirred with a solution of NaHCO3 (0.84 g, 10 mmol) in water (10 mL) for 2 h. The organic layer was separated, the aqueous layer was extracted with chloroform (20 mL), and the combined organic solutions were evaporated in vacuum. Recrystallisation of the residue from heptane/benzene (3:1) mixture afforded 1.32 g (75%) of compound 8a with m.p. 121–124 °C and [α]25D −55.0° (c 1.31, CHCl3). Found, %: C 41.25, H 7.56, N 7.80, S 9.03. C12H26N2O4SSi2. Calculated, %: C 41.11, H 7.48, N 7.99, S 9.15. IR spectrum (KBr, ν, cm−1): 1631 s (CO), 1325 s, 1148 s (SO2). 1H NMR spectrum (CDCl3, δ, ppm): 0.18, 0.19, 0.21 and 0.31 (four s, 12H, 2Si(CH3)2); 1.88–2.34 (m, 4H, C3H2 and C4H2 Pro); 2.7 and 3.42 (dd, 2H, NCH2Si, 3JHH 15.34 Hz); 2.83 (dd, 2H, NCH2Si, 3JHH 15.34 Hz); 3.01 (s, 3H, SCH3); 3.45–3.52 and 3.56–3.63 (two m, 2H, C5H2 Pro); 4.80–4.87 (m, 1H, C2H Pro). 13C NMR spectrum (CDCl3, δ, ppm): –0.76 ÷ 0.00 (m, 2SiMe2); 24.65 (4C Pro); 30.79 (3C Pro); 39.9 (SC); 38.1 and 40.29 (two s, NCH2Si); 47.54 (5C Pro); 58.92 (C2 Pro); 169.64 (CO). 29Si NMR spectrum (CDCl3, δ, ppm): 8.0, 10.5.
2,2,6,6-Tetramethyl-4-[N-tosyl-(S)-prolinyl]-2,6-disilamorpholine (8b)
Prepared similar to 8a from 1.34 g of 6b. Yield 1.66 g (78%) with m. p. 110–112 °C (from heptane–benzene, 10:1) and [α]25D −2.92° (c 1.85, CHCl3). Found, %: C 50.51, H 7.24, N 6.62, S 7.45. C18H30N2O4SSi2. Calculated, %: C 50.67, H 7.09, N 6.57, S 7.52. IR spectrum (KBr, ν, cm−1): 1629 s (CO), 1580 m (Ar), 1325 s, 1148 s (SO2). 1H NMR spectrum (CDCl3, δ, ppm): 0.17, 0.21, 0.23 and 0.34 (four s, 12H, 2Si(CH3)2); 1.88–2.05 (m, 4H, C3H2 and C4H2 Pro); 2.42 (s, 3H, ArCH3); 2.95 and 3.14 (dd, 2H, NCH2Si, 3JHH 15.0 Hz); 2.99 and 3.04 (dd, 2H, NCH2Si, 3JHH 15.95 Hz); 3.39–3.46 and 3.50–3.57 (two m, 2H, C5H2 Pro); 4.87–4.92 (m, 1H, C2H Pro); 7.28 and 7.79 (two d, 4H, Ar, 3JHH 8 Hz). 13C NMR spectrum (CDCl3, δ, ppm): –0.57 ÷ 0.00 (m, 2SiMe2); 21.47 (Me); 24.75 (4C Pro); 30.89 (3C Pro); 37.96 and 40.41 (two s, NCH2Si); 48.09 (5C Pro); 57.60 (C2 Pro); 127.48 (C2 and C6 Ar), 129.30 (C3 and C5 Ar), 136.50 (C1 Ar), 143.04 (C4 Ar), 169.52 (CO). 29Si NMR spectrum (CDCl3, δ, ppm): 7.9, 10.4.
2,2,6,6-Tetramethyl-4-(N-mesylsarcosinyl)-2,6-disilamorpholine (8c)
Prepared similar to 8a from 0.83 g of 6c. Yield 1.3 g (80%) with m.p. 151–153 °C (from heptane–benzene, 7:1). Found, %: C 37.28, H 7.24, N 8.64, S 9.51. C10H24N2O4SSi2. Calculated, %: C 37.01, H 7.45, N 8.63, S 9.88. IR spectrum (KBr, ν, cm−1): 1628 s (CO), 1323 s, 1153 s (SO2). 1H NMR spectrum (CDCl3, δ, ppm): 0.18 and 0.23 (two s, 12H, 2Si(CH3)2); 2.79 and 3.06 (two s, 4H, NCH2Si); 2.98 (s, 3H, NCH3); 2.99 (s, 3H, SCH3); 4.13 (s, 2H, NCH2). 13C NMR spectrum (CDCl3, δ, ppm): –0.37 and −0.19 (two s, 2Si(CH3)2); 35.44 (NMe); 37.95 and 39.70 (two s, NCH2Si); 38.15 (SMe); 51.56 (NCCO); 165.58 (CO). 29Si NMR spectrum (CDCl3, δ, ppm): 8.3, 10.4.
N′,N′-Bis(dimethylfluorosilylmethyl)-N-mesyl-(S)-prolinamide (9a)
Boron trifluoride (0.36 g, 2.5 mmol) was added dropwise to a solution of 8a (0.88 g, 2.5 mmol) in acetonitrile (5 mL). The reaction mixture was refluxed for 2 h, then evaporated in vacuum. The remaining oil was refluxed with benzene (15 mL), the precipitate was filtered out, and the solution was evaporated in vacuum. The residue was recrystallised from heptane to afford 0.76 g (82%) of 9a with m.p. 100–101 °C. Found, %: C 34.61, H 6.88, N 7.86, S 8.92. C10H24F2N2O3SSi2. Calculated, %: C 34.66, H 6.98, N 8.08, S 9.25. IR spectrum (KBr, ν, cm−1): 1610 s, 1505 w (CO), 1319 s, 1134 s (SO2). 1H NMR spectrum (CDCl3, δ, ppm): 0.22 and 0.31 (two s, 6H, SiV(CH3)2); 0.41 and 0.46 (dd, 6H, SiIV(CH3)2, 3JHF 7.67 Hz); 1.86–1.93, 2.03–2.18 and 2.28–2.36 (m, 4H, C3H2 and C4H2 Pro); 2.44 and 2.59 (dd, 2H, NCH2SiV, 3JHH 15.74 Hz); 2.98 (s, 3H, SCH3); 3.00–3.05 and 3.25–3.30 (two m, 2H, NCH2SiIV); 3.44–3.5 and 3.58–3.63 (two m, 2H, C5H2 Pro); 4.75–4.77 (m, 1H, C2H Pro). 13C NMR spectrum (CDCl3, δ, ppm): −1.9 ÷ −1.7 (m, SiIVCH3); 1.1–1.7 (m, SiVCH3); 24.95 (C4 Pro); 30.93 (C3 Pro); 39.23 (SC); 41.1 ÷ 41.5 (m, CH2SiV and CH2SiIV); 47.72 (C5 Pro); 56.59 (C2 Pro); 172.51 (CO). 19F NMR spectrum (CDCl3, δ, ppm): −159.15; −121.88. 29Si NMR spectrum (CDCl3, δ, ppm): −15.5 (d, 1JSiF 252 Hz), 28.9 (d, 1JSiF 284 Hz). 29Si NMR CP/MAS spectrum (δ, ppm): −37.2 (d, 1JSiF 880 Hz), 32.7 (d, 1JSiF 1024 Hz).
N′,N′-Bis(dimethylfluorosilylmethyl)-N-tosyl-(S)-prolinamide (9b)
Prepared similar to 9a from 1.1 g of 8b. Yield 0.9 g (80%) with m. p. 87–88 °C (from heptane). Found, %: C 48.23, H 6.80, N 6.15, S 7.20. C18H30F2N2O3SSi2. Calculated, %: C 48.18, H 6.74, N 6.24, S 7.15. IR spectrum (KBr, ν, cm−1): 1602 s, 1515 w (CO), 1336 s, 1151 s (SO2). 1H NMR spectrum (CDCl3, δ, ppm): 0.18 and 0.22 (two s, 6H, SiV(CH3)2); 0.41–0.44 (m, 6H, SiIV(CH3)2); 1.83–1.93 and 2.05–2.16 (two m, 4H, C3H2 and C4H2 Pro); 2.43 and 2.51 (dd, 2H, NCH2SiV, 3JHH 15.74 Hz); 2.45 (s, 3H, ArCH3); 3.04–3.08 and 3.52–3.59 (two m, 2H, NCH2SiIV); 3.35–3.4 and 3.46–3.52 (two m, 2H, C5H2 Pro); 4.78–4.81 (m, 1H, C2H Pro), 7.32 (d, 2H, Ar, 3JHH 8.07 Hz), 7.75 (d, 2H, Ar, 3JHH 8.07 Hz). 13C NMR spectrum (CDCl3, δ, ppm): −1.92 ÷ −1.78 (m, SiIVCH3); 1.20–1.37 (m, SiVCH3); 21.46 (ArMe); 24.89 (C4 Pro); 30.63 (C3 Pro); 41.27 (d, CH2SiIV, 3JCF 16.69 Hz); 41.36–41.47 (m, CH2SiV); 48.24 (C5 Pro); 55.45 (C2 Pro); 127.31 (C2 and C6 Ar), 129.64 (C3 and C5 Ar), 135.89 (C1 Ar), 143.74 (C4 Ar); 172.39 (CO). 19F NMR spectrum (CDCl3, δ, ppm): −159.47; −119.31. 29Si NMR spectrum (CDCl3, δ, ppm): −19.1 (d, 1JSiF 236 Hz), 28.1 (d, 1JSiF 276 Hz). 29Si NMR CP/MAS spectrum (δ, ppm): −32.0 (d, 1JSiF 251 Hz), 30.7 (d, 1JSiF 292 Hz).
N′,N′-Bis(dimethylfluorosilylmethyl)-N-mesylsarcosinamide (9c)
Prepared similar to 9a from 0.80 g of 8c. Yield 0.73 g (85%) with m. p. 135–136 °C (from heptane). Found, %: C 34.61, H 6.88, N 7.86, S 8.92. C10H24F2N2O3SSi2. Calculated, %: C 34.66, H 6.98, N 8.08, S 9.25. IR spectrum (KBr, ν, cm−1): 1610 s, 1505 w (CO), 1319 s, 1134 s (SO2). 1H NMR spectrum (CDCl3, δ, ppm): 0.30 (two s, 6H, Si(CH3)2); 0.39–0.43 (m, 6H, SiIV(CH3)2); 2.56 and 3.05 (two s, 4H, NCH2Si); 2.96 (s, 3H, NCH3); 2.98 (s, 3H, SCH3); 4.17 (s, 2H, NCH2). 13C NMR spectrum (CDCl3, δ, ppm): −1.8 (d, SiMe2, 2JCF 14.5 Hz), 1.2 (s, SiMe2); 35.3 (NMe); 37.9 and 40.8 (two s, NCH2Si); 41.4 (SMe); 49.7 (NCCO); 168.6 (CO). NMR 19F spectrum (CDCl3, δ, ppm): −159.15; −125.46. 29Si NMR spectrum (CDCl3, δ, ppm): −10.5 (d, 1JSiF 248 Hz), 29.2 (d, 1JSiF 287 Hz).
Single crystals suitable for X-ray diffraction analysis were obtained by recrystallisation from: orthorhombic 8a—heptane/benzene 3:1; monoclinic 8a′—ethanol; 8c—heptane/benzene 7:1; 9a—heptane. X-ray diffraction measurements were carried out using Bruker Smart 1000 CCD and Bruker Smart Apex II CCD diffractometers at 100 K. The frames were integrated using SMART and APEX2 program packages.28 The correction for absorption was made using SADABS program.29 The details of crystallographic data and experimental conditions are given in Table 5.
| 8a | 8a′ | 8c | 9a | |
|---|---|---|---|---|
| Molecular formula | C12H26N2O4SSi2 | C12H26N2O4SSi2 | C10H24N2O4SSi2 | C12H26F2N2O3SSi2 |
| Formula weight | 350.59 | 350.59 | 324.55 | 372.59 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P212121 | P21 | P21/n | P212121 |
| Flack parameter | 0.027(18) | 0.03(9) | — | 0.016(13) |
| Z | 8 | 2 | 4 | 8 |
| a, Å | 9.6000(5) | 7.436(4) | 15.1089(9) | 13.0441(9) |
| b, Å | 14.2422(7) | 9.474(6) | 6.6275(4) | 15.9282(11) |
| c, Å | 27.0844(14) | 14.125(9) | 16.4442(10) | 18.2980(13) |
| α, ° | 90 | 90 | 90 | 90 |
| β, ° | 90 | 104.082(9) | 98.7620(10) | 90 |
| γ, ° | 90 | 90 | 90 | 90 |
| V, Å3 | 3703.1(3) | 965.2(10) | 1627.41(17) | 3801.8(5) |
| ρ calc (g cm−3) | 1.258 | 1.206 | 1.325 | 1.302 |
| μ, cm−1 | 3.19 | 3.06 | 3.57 | 3.25 |
| F(000) | 1504 | 376 | 696 | 1584 |
| 2θmax, ° | 61.03 | 60.22 | 60.06 | 61.06 |
| Reflections collected | 50072 |
10584 |
30094 |
65941 |
| Independent reflections (Rint) | 11308 (0.0355) |
5331 (0.00) | 4740 (0.0311) | 11597 (0.0327) |
| Number of reflections with I > 2σ(I) | 10588 |
3228 | 3981 | 10557 |
| Parameters | 389 | 195 | 178 | 411 |
| R 1 [I > 2σ(I)] | 0.0344 | 0.0579 | 0.0484 | 0.0266 |
| wR2 (all independent reflections) | 0.0793 | 0.1168 | 0.1022 | 0.0698 |
| GOF | 1.071 | 1.000 | 1.034 | 1.084 |
| ρ min/ρmax (e Å−3) | 0.556/−0.323 | 0.873/−0.494 | 0.565/−0.358 | 0.381/−0.197 |
The structures were solved by the direct method by XS program30 and refined by full-matrix least-squares technique against F2 in the anisotropic–isotropic approximation using XL program.30 Atom H20 in s was located from the difference Fourier maps and refined freely. All remaining hydrogen atoms were placed in geometrically calculated positions and refined in rigid body model (Uiso(H) = 1.2Ueq(CH, CH2), Uiso(H) = 1.5Ueq(CH3)). The Flack parameter confirms (S)-configuration of the proline fragment. Preparation of graphic materials was performed using OLEX2 software package.31 Crystallographic data for the structural analysis of 8a, 8a′, 8c and 9a have been deposited with the Cambridge Crystallographic Data Centre (CCDC nos 1059570–1059573).
Acknowledgements
This work was carried out as a part of the research activities of the Science and Education Centre for the Synthesis and Investigation of Biologically Active Compounds at the N. I. Pirogov Russian National Research Medical University and supported by the Russian Foundation for Basic Research (grants nos 16-03-00957, 16-33-60168 and 16-33-00956).References
- (a) C. Chuit, R. J. P. Corriu, C. Reye and J. C. Young, Chem. Rev., 1993, 93, 1371–1448 Search PubMed; (b) D. Kost and I. Kalikhman, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, Chichester, U.K., 1998, vol. 2, pp. 1339–1445 Search PubMed; (c) J. Wagler, U. Böhme and E. Kroke, in Functional Molecular Silicon Compounds I: Regular Oxidation States, ed. D. Scheschkewitz, Springer, 2014, pp. 29–105 Search PubMed.
- (a) V. V. Negrebetsky and Y. I. Baukov, Russ. Chem. Bull., 1997, 46, 1807–1831 CrossRef CAS; (b) V. V. Negrebetsky, S. N. Tandura and Y. I. Baukov, Russ. Chem. Rev., 2009, 78, 21–51 CrossRef CAS; (c) A. A. Nikolin and V. V. Negrebetsky, Russ. Chem. Rev., 2014, 83(9), 848–883 CrossRef CAS.
- A. Chandra, R. Sheker, Z. Chen, T. Hatanaka, T. Minami and Y. Hatanaka, Organometallics, 2013, 32, 3575–3582 CrossRef CAS; Y. Li, C. deKock, P. J. Smith, H. Guzgay, D. T. Hendricks, K. Naran, V. Mizrahi, D. F. Warner, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 141–150 CrossRef; T. Baramov, K. Keijzer, E. Irran, E. Mösker, M.-H. Baik and R. Süssmuth, Chem.–Eur. J., 2013, 19, 10536–10542 CrossRef PubMed; Y. Tokoro, H. Yeo, K. Tanaka and Y. Chujo, Polym. Chem., 2013, 4, 5237–5242 RSC; Y. Hatanaka, S. Okada, T. Minami, M. Goto and K. Shimada, Organometallics, 2005, 24, 1053–1055 CrossRef.
- E. Lukevics and L. Ignatovich, in Metallotherapeutic Drugs and Metal-Based Diagnostic Agents, ed. M. Gielen and T. E. R. Tiekink, Wiley, 2005, pp. 83–108 Search PubMed.
- (a) K. Junold, J. A. Baus, C. Burschka, D. Auerhammer and R. Tacke, Chem.–Eur. J., 2012, 18, 16288–16291 CrossRef CAS PubMed; (b) K. Junold, J. A. Baus, C. Burschka, C. F. Guerra, F. M. Bickehaupt and R. Tacke, Chem.–Eur. J., 2014, 20, 12411–112415 CrossRef CAS PubMed; (c) S. Metz, C. Burschka, D. Platte and R. Tacke, Angew. Chem., Int. Ed., 2007, 46, 7006–7009 CrossRef CAS PubMed; (d) D. Troegel, C. Burschka, S. Riedel, M. Kaupp and R. Tacke, Angew. Chem., Int. Ed., 2007, 46, 7001–7005 CrossRef CAS PubMed; (e) R. Tacke, C. Burschka, I. Richter, B. Wagner and R. Willeke, J. Am. Chem. Soc., 2000, 122, 8480–8485 CrossRef CAS.
- (a) R. W. Hillyard Jr, M. C. Ryan and C. H. Yoder, J. Organomet. Chem., 1978, 153(3), 369–377 CrossRef; (b) K. D. Onan, A. T. McPhail, C. H. Yoder and R. W. Hillyard Jr, Chem. Commun., 1978, 5, 209–210 RSC; (c) A. R. Bassindale and M. Borbaruah, J. Chem. Soc., Chem. Commun., 1993, 352–353 RSC; (d) A. G. Shipov, E. P. Kramarova, E. A. Mamaeva, O. A. Zamyshlyaeva, V. V. Negrebetsky, Y. E. Ovchinnikov, S. A. Pogozhikh, A. R. Bassindale, P. G. Taylor and Y. I. Baukov, J. Organomet. Chem., 2001, 620, 139–145 CrossRef CAS; (e) A. A. Korlyukov, K. A. Lyssenko, M. Y. Antipin, A. G. Shipov, O. A. Zamyshlyaeva, E. P. Kramarova, V. V. Negrebetsky, S. A. Pogozhikh, Y. E. Ovchinnikov and Y. I. Baukov, Russ. Chem. Bull., 2004, 53(9), 1924–1931 CrossRef CAS.
- (a) A. A. Nikolin, E. P. Kramarova, A. G. Shipov, Y. I. Baukov, V. V. Negrebetsky, A. A. Korlyukov, D. E. Arkhipov, A. Bowden, S. Y. Bylikin, A. R. Bassindale and P. G. Taylor, Organometallics, 2012, 31(14), 4988–4997 CrossRef CAS; (b) A. A. Nikolin, O. V. Kuznetsova, D. E. Arkhipov, E. P. Kramarova, A. G. Shipov, A. N. Egorochkin, A. A. Korlyukov, Y. I. Baukov and V. V. Negrebetsky, Russ. Chem. Bull., 2013, 8, 1892–1899 CrossRef.
- (a) E. P. Kramarova, V. V. Negrebetsky, A. G. Shipov and Y. I. Baukov, Russ. J. Gen. Chem., 1994, 64 ( Zh. Obshch. Khim , 1994 , 64 , 1222–1223 ) CAS; (b) Y. I. Baukov, A. G. Shipov, E. P. Kramarova, E. A. Mamaeva, O. A. Zamyshlyaeva, N. A. Anisimova and V. V. Negrebetsky, Russ. J. Org. Chem., 1996, 32, 1259–1271 CAS.
- Y. I. Baukov, E. P. Kramarova, A. G. Shipov, G. I. Oleneva, O. B. Artamkina, A. I. Albanov, M. G. Voronkov and V. A. Pestunovich, J. Gen. Chem. USSR, 1989, 59 ( Zh. Obshch. Khim , 1989 , 59 , 127–145 ) CAS.
- A. A. Nikolin, D. E. Arkhipov, A. G. Shipov, E. P. Kramarova, N. A. Koval'chuk, A. A. Korlyukov, V. V. Negrebetsky, Y. I. Baukov, A. R. Bassindale, P. G. Taylor, A. Bowden and S. Y. Bylikin, Chem. Heterocycl. Compd., 2012, 47(12), 1565–1583 CrossRef CAS.
- E. P. Doronina, V. F. Sidorkin and N. F. Lazareva, J. Phys. Chem. A, 2015, 119, 3663–3673 CrossRef CAS PubMed.
- N. N. Chipanina, T. N. Aksamentova, M. G. Voronkov and V. K. Turchaninov, J. Struct. Chem., 2006, 46, 1066–1070ss CrossRef.
- V. V. Negrebetsky, A. G. Shipov, E. P. Kramarova, V. V. Negrebetsky and Y. I. Baukov, J. Organomet. Chem., 1997, 530, 1–12 CrossRef CAS.
- O. B. Bannikova, A thesis for the degree of candidate of chemical Sciences, The Institute of Organic Chemistry, Russian Academy of Science, Irkutsk, 1986, p. 20.
- (a) A. Lends, E. Olszewska, S. Belyakov, N. Erchak and E. Liepinsh, Heteroat. Chem., 2015, 26, 12–28 CrossRef CAS; (b) R. Tacke, J. Becht, O. Dannappel, R. Ahlrichs, R. Schneider, W. S. Sheldrick, J. Hahn and F. Kiesgen, Organometallics, 1996, 15, 2060–2077 CrossRef CAS; (c) O. Girdhberg, I. Kalikhman, D. Stalke, B. Walfort and D. Kost, J. Mol. Struct., 2003, 661–662, 259–264 CrossRef; (d) V. V. Negrebetsky, S. Y. Bylikin, A. G. Shipov, Y. I. Baukov, A. R. Bassindale and P. G. Taylor, J. Organomet. Chem., 2003, 678, 39–47 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- V. F. Sidorkin, E. F. Belogolova and V. A. Pestunovich, Chem.–Eur. J., 2006, 12, 2021–2031 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, C.01 Search PubMed.
- G. A. Zhurko and D. A. Zhurko, Chemcraft Program, Academic version 1.7, 2011 Search PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2016, 44, D1202–D1213, DOI:10.1093/nar/gkv951.
- D. A. Filimonov, A. A. Lagunin, T. A. Gloriozova, A. V. Rudik, D. S. Druzhilovskii, P. V. Pogodin and V. V. Poroikov, Chem. Heterocycl. Compd., 2014, 50, 444–457 CrossRef CAS.
- A. Lagunin, A. Zakharov, D. Filimonov and V. Poroikov, Mol. Inf, 2011, 30, 241–250 CrossRef CAS PubMed.
- Pulse methods in 1D and 2 D liquid-phase NMR, ed. W. S. Brey, Academic Press, New York, 1988, p. 561 Search PubMed.
- A. L. van Geet, Analyt. Chem., 1970, 42, 679–680 CrossRef CAS.
- G. Haegele, R. Fuhler and T. Lenzen, Comp. Chem., 1995, 19, 277–282 CrossRef.
- G. Binsch, in Dynamic Nuclear Magnetic Resonance Spectroscopy, ed. L. Jackman and M. F. A. Cotton, Academic Press, New York, 1975, p. 45 Search PubMed.
- J. Tesse, Bull. Soc. Chim. Fr., 1973, 787–793 CAS.
- APEX2, SMART, SAINT, SAINT-Plus, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1059570–1059573. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra14450k |
| This journal is © The Royal Society of Chemistry 2016 |