
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Real-time broadband proton-homodecoupled CLIP/CLAP-HSQC for automated measurement of heteronuclear one-bond coupling constants†
István
Timári
a,
Lukas
Kaltschnee
b,
Mária H.
Raics
a,
Felix
Roth
b,
Nicholle G. A.
Bell
c,
Ralph W.
Adams
 d,
Mathias
Nilsson
d,
Dušan
Uhrín
c,
Gareth A.
Morris
d,
Christina M.
Thiele
b and
Katalin E.
Kövér
*a
d,
Mathias
Nilsson
d,
Dušan
Uhrín
c,
Gareth A.
Morris
d,
Christina M.
Thiele
b and
Katalin E.
Kövér
*a
aDepartment of Inorganic and Analytical Chemistry, University of Debrecen, Egyetem tér 1, H-4032 Debrecen, Hungary. E-mail: kover@science.unideb.hu
bClemens-Schöpf-Institut für Organische Chemie und Biochemie, Technische Universität Darmstadt, Alarich-Weiss-Straße 4, D-64287 Darmstadt, Germany
cEastCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK
dSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 2nd September 2016
Abstract
A new method is proposed that allows broadband homonuclear decoupled CLIP/CLAP-HSQC NMR spectra to be acquired at virtually no extra cost in measurement time. The real-time (windowed) acquisition protocol applied follows a scheme recently devised for recording pure shift (broadband homonuclear decoupled) heteronuclear single quantum correlation (HSQC) spectra. To minimize systematic errors in the apparent coupling constants obtained using real-time homonuclear decoupling, we extended the acquisition scheme to include cycling of radiofrequency pulse phases both from chunk to chunk during windowed acquisition, and from scan to scan during time averaging, allowing robust coupling constant measurement. The new real-time pure shift CLIP/CLAP-HSQC experiments are designed to speed up coupling constant determination, to increase the sensitivity of measurement in favorable cases, and to simplify the extraction of accurate one-bond heteronuclear couplings from pure in- or anti-phase doublets using automatic peak picking. The scope and limitations of the method are discussed, and a variety of experimental tests are reported.
Introduction
CLIP/CLAP-HSQC (CLean In-Phase/CLean Anti-Phase HSQC) experiments1 and their variants2 are some of the most widely used NMR methods for measuring heteronuclear couplings (1JXH) between directly bonded X–1H spin pairs. In conventional CLIP/CLAP-HSQC spectra,1 however, the X-coupled F2 doublets retain the complex multiplet structures caused by proton–proton couplings, reducing the spectral resolution and, along with that, the precision of coupling constant measurement.To circumvent this limitation of conventional experiments, recently-proposed pure shift (PS) CLIP/CLAP-HSQC methods3–5 provide a practical remedy, giving resolution improvements of close to an order of magnitude. These pure shift methods use interferogram-based acquisition,6,7 enabling the acquisition of spectra of high spectral quality but requiring significantly longer experiments than the corresponding conventional techniques without homodecoupling.
To reduce the duration of pure shift HSQC experiments, it has recently been shown8,9 that it is possible to acquire decoupled signals using real-time acquisition, in a single free induction decay during which homonuclear proton–proton coupling evolution is periodically refocused by applying a BIRD (BIlinear Rotation Decoupling) pulse sequence element10 in combination with a non-selective 180° proton pulse. The Zangger–Sterk11 and HOBS (HOmonuclear Band-Selective)/BASH (Band-Selective Homonuclear)12–14 pulse sequence elements for homonuclear decoupling have also been combined with this real-time, or “instant”15 acquisition strategy. The great advantage of real-time pure shift methods is that, in contrast to interferogram-based pure shift acquisition, they incur no extra experiment time penalty.16–18 Indeed, in favorable circumstances real time pure shift methods can, by virtue of collapsing complex multiplets into strong single signals, show better sensitivity than their conventional analogs.9
While real-time BASH decoupling does not cause sensitivity loss, it is not broadband, only providing homonuclear decoupling for a defined spectral region that does not contain mutually J-coupled protons (such as the amide region in protein spectra).12–14 In heteronuclear one-bond correlation experiments, the BIRD method is broadband and also retains full sensitivity: since in HSQC experiments only those protons directly coupled to X (e.g.13C, 15N) are being detected, there is no extra signal loss when BIRD sequence elements are used.9 We have recently compared in detail the performance of BIRD and BASH-decoupled 1H–15N HSQC methods, including the effectiveness of water signal suppression, in protein studies.19 Here we demonstrate that it is possible to implement a real-time pure shift acquisition scheme devised for variants of HSQC9,19,20 in CLIP/CLAP-HSQC pulse sequences with preservation of the one-bond heteronuclear coupling. The use of real-time decoupling with simultaneous one-bond coupling measurement in the direct dimension is significantly less straightforward than it might at first sight seem, because of the potential for small systematic errors in successive J-refocusing pulse sequence elements to accumulate during acquisition, leading to slightly distorted spectra and to small but unwelcome errors in the one-bond coupling constants measured. It will be shown that such errors can be kept within acceptable limits by careful sequence design, with the choice of successive pulse phases during acquisition being particularly important.
Results and discussion
As in the real-time pure shift HSQC experiment,9 trains of isotope-selective BIRD modules combined with non-selective 1H 180° pulses are used here to refocus nJHH modulation midway through each of the acquired data chunks (n ≥ 3, here). The combined effect of the BIRD element and the hard 180° proton pulse is to leave protons directly coupled to X (e.g.13C, 15N), whose signals are detected, unaffected, but to invert protons not directly coupled to X. Because CLIP/CLAP-HSQC experiments do not use broadband X decoupling during acquisition, in order to retain the one-bond couplings (1JXH) it is necessary here to use the BIRD(d) module,21 which differs from the parent BIRD(d,X) module in having an extra 180° X pulse added so that the X spin is not left inverted. In the real-time BIRD(d) CLIP-HSQC sequence of Fig. 1 – as in most other HSQC experiments – a refocusing spin-echo element using a simultaneous 1H/X 180° pulse pair at its midpoint is included before acquisition. Here, to generate pure in-phase magnetization for coupling constant measurement, an extra 90° X purging pulse is applied prior to the start of real-time PS acquisition.1 In contrast, in the pure shift CLAP-HSQC experiment the greyed 180° and 90° X pulses in Fig. 1 are discarded to retain anti-phase magnetization – generated by the X → 1H coherence transfer step – for real-time acquisition of the X-coupled but proton-homodecoupled signal. The resultant PS CLIP/CLAP-HSQC spectra provide broadband proton-homodecoupled pure in- or anti-phase X-coupled doublets with significantly enhanced resolution, allowing direct and highly precise measurement of heteronuclear one-bond couplings.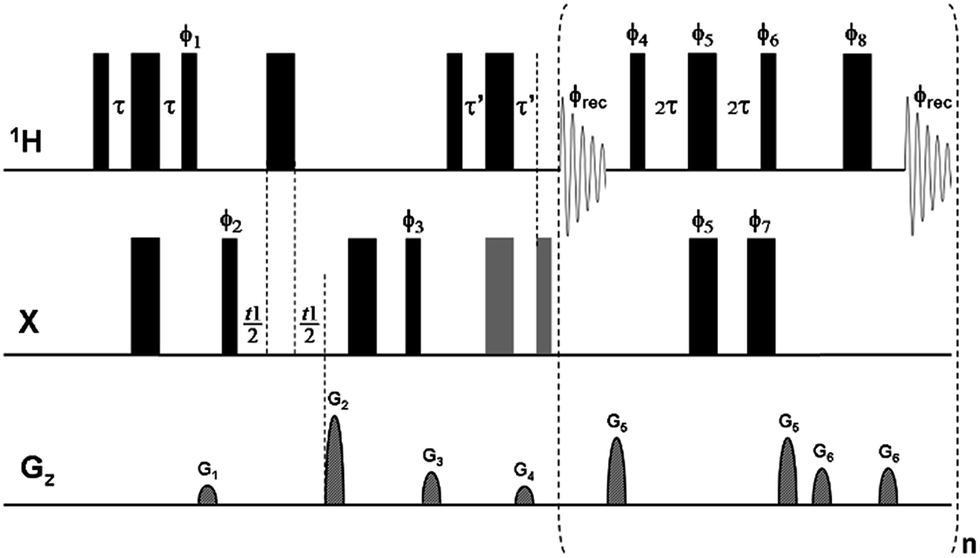 | ||
Fig. 1 Pulse sequence scheme for real-time broadband proton-homodecoupled CLIP/CLAP-HSQC experiments. Narrow and wide filled bars correspond to 90° and 180° pulses, respectively, with phase x unless indicated otherwise. In the CLIP-HSQC sequence additional carbon 180° and 90° pulses (shown in gray) are employed to refocus antiphase proton magnetization and to remove undesired residual dispersive components respectively prior to detection. These pulses are omitted in the CLAP-HSQC experiment, where the antiphase proton magnetization is retained and detected. Delays are set as follows: τ = τ′ = 1/(4 × 1JXH). Coherence order selection and echo–antiecho phase sensitive detection in the X-dimension are achieved with gradient pulses G2 and G4 in the ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20.1 for 13C and 80:8.1 for 15N, respectively. Purging gradient pulses G1 and G3 are typically 1 ms duration (p16) followed by a recovery delay (d16) of 100 μs. Each J-refocusing block applied during acquisition consists of a BIRD(d) element, a hard 180° proton pulse, and a data acquisition window. The first and last chunks are half the duration (AQ/2n) of the remaining chunks (AQ/n). CTP selection gradient pulses G5 and G6 are used around the BIRD(d) block and the hard 180° proton pulse between data chunks, with 0.5 ms duration and 100 μs recovery delay. Phase cycles are ϕ1 = y; ϕ2 = (x)2, (−x)2; ϕ3 = (x)4, (−x)4; ϕ4 = x; ϕ5 = y, −y; ϕ6 = −x; ϕ7 = x, −x; ϕ8 = x, −x; and ϕrec = (x)2, (−x)4, (x)2. Phase sequencing of J-refocusing elements: ϕchunk-to-chunk(n) = {(x)4, (−x)6, (x)4, (−x)2}runs over n; ϕ4 = x + ϕchunk-to-chunk(n); ϕ5 = y, −y + ϕchunk-to-chunk(n); ϕ6 = −x + ϕchunk-to-chunk(n); ϕ7 = x, −x + ϕchunk-to-chunk(n); ϕ8 = x, −x + ϕchunk-to-chunk(n). For the explicit phase sequences and cycles see Table S2.† :20.1 for 13C and 80:8.1 for 15N, respectively. Purging gradient pulses G1 and G3 are typically 1 ms duration (p16) followed by a recovery delay (d16) of 100 μs. Each J-refocusing block applied during acquisition consists of a BIRD(d) element, a hard 180° proton pulse, and a data acquisition window. The first and last chunks are half the duration (AQ/2n) of the remaining chunks (AQ/n). CTP selection gradient pulses G5 and G6 are used around the BIRD(d) block and the hard 180° proton pulse between data chunks, with 0.5 ms duration and 100 μs recovery delay. Phase cycles are ϕ1 = y; ϕ2 = (x)2, (−x)2; ϕ3 = (x)4, (−x)4; ϕ4 = x; ϕ5 = y, −y; ϕ6 = −x; ϕ7 = x, −x; ϕ8 = x, −x; and ϕrec = (x)2, (−x)4, (x)2. Phase sequencing of J-refocusing elements: ϕchunk-to-chunk(n) = {(x)4, (−x)6, (x)4, (−x)2}runs over n; ϕ4 = x + ϕchunk-to-chunk(n); ϕ5 = y, −y + ϕchunk-to-chunk(n); ϕ6 = −x + ϕchunk-to-chunk(n); ϕ7 = x, −x + ϕchunk-to-chunk(n); ϕ8 = x, −x + ϕchunk-to-chunk(n). For the explicit phase sequences and cycles see Table S2.† | ||
One challenge that such real-time homodecoupling techniques present is that they require careful design if clean results are to be obtained. The central problem, spectral perturbations caused by the cumulative effect of successive imperfect pulse sequence elements, is one that is common to all experiments in which windowed acquisition, alternating spin manipulations and chunks of data acquisition, is used. In high resolution NMR this includes real-time heteronuclear J-scaling22 (for which a detailed analysis of the effects of pulse imperfections has been presented),23 real-time pure shift NMR,8,9,12,14,15,19,24 and J-up- and down-scaling.25–27 With one honorable exception,14 such problems have largely been ignored in the literature.
To take a simple example, consider a real-time experiment in which an imperfect pulse sequence element is applied at intervals τ during data acquisition. Possible (and indeed unavoidable) sources of imperfection include B1 inhomogeneity, resonance offset, and (in BIRD-based experiments) J mismatch. Suppose that the imperfect element is equivalent to the ideal spin manipulation required plus a small extra flip angle α radians. The spins will see the average Hamiltonian sought, plus a periodic series of unwanted pulses that correspond in the frequency domain to a comb of very weak irradiations of amplitudes α/τ rad s−1 at the transmitter offset plus and minus integer multiples of 1/τ Hz.28 If, as will often be the case, these sidebands do not fall close to any resonance, they will be too weak to have any significant effect on the spectrum. If, however, one of the sidebands is close to or coincident with a spectral line, that line will be perturbed, for example by being split and/or slightly displaced. In many experiments, for example pure shift NMR, such effects can safely be neglected, but where, as here, accurate coupling constant measurements are required, precautions must be taken to minimize the impact of periodic errors on real-time measurements.
Two obvious strategies for reducing the impact of pulse imperfections in real-time windowed acquisition are to minimize the cumulative effects of imperfect pulses by manipulating the phases of successive pulse sequence elements during acquisition of a single transient;14 and to average out the perturbations that remain by manipulating those phases from transient to transient during time averaging. We refer here to the former as a phase sequence (within a single transient) and to the latter, as is customary, as a phase cycle (from transient to transient).
To assess the utility of the proposed real-time BIRD(d) CLIP/CLAP-HSQC sequences, tests of robustness with respect to proton resonance offset, J mismatch (BIRD/INEPT delay settings) and pulse (mis)calibration were made using a model sample of 0.345 M methyl α-D-mannopyranoside in D2O. The one-bond 1H–13C coupling constant of the CH3-group measured directly from the 13C-satellites in the proton spectrum (1JCH = 144.06 ± 0.02 Hz) was used as a reference for testing the accuracy of the experiments proposed. The phase sequence of J-refocusing elements (BIRD(d) and 180° proton pulse) applied during acquisition and the scan to scan phase cycle were optimized empirically by testing various phase combinations (see details in Table S1 of the ESI†). The sequence and cycle which gave the best accuracy and the least sensitivity to variation of the proton resonance offset and duration of the BIRD/INEPT delay (2τ, τ) are given in the legends of Fig. 1 and in Table S2.†
To test the robustness of the phase program optimized sequences of Fig. 1 with respect to proton resonance offset, the spectrometer operating (carrier) frequency was varied in two frequency ranges: from 700 Hz up to 780 Hz off-resonance from the CH3-signal, and from 1900 Hz up to 1980 Hz off-resonance, both in steps of 2 Hz, to give 40 experiments in each case. The latter offset corresponds to half of an average protein NH-frequency window at 800 MHz. The 40 spectra recorded with the optimized phase program yielded an average value of 1JCH = 143.96 Hz with a maximum deviation of 0.42 Hz and a standard deviation (σJ) of 0.17 Hz in the 700–780 Hz offset range, and 1JCH = 144.04 Hz with a maximum deviation of 1.05 Hz and σJ of 0.40 Hz in the 1900–1980 Hz range (see Fig. S1A and B† for corresponding plots of apparent J vs. offset). As expected, the uncertainty in 1JCH increases with offset, so it can be helpful to make measurements with different carrier frequencies for best results. Fig. S2† shows overlays of the 40 antiphase CH3-doublets measured using the CLAP variant of the sequence of Fig. 1 for the two offset ranges.
The impact of the duration of the BIRD/INEPT delays (2τ, τ) was also tested, with τ set for the range of 1JCH from 125 Hz to 175 Hz, corresponding to a typical spread of coupling constants in samples dissolved in weakly orienting media for RDC (Residual Dipolar Coupling) measurement. Using τ values set for a range of 15 Hz either side of the actual 1JCH value of 144 Hz gave a maximum deviation of 0.6 Hz, with τ values at the edges of the range tending to overestimate 1JCH. As τ was moved further from the optimum value the latter tendency increased, with errors reaching 1.4 Hz at ±30 Hz (see experimental data in Table S3 of the ESI†). The proposed experiment is thus reasonably tolerant of variation in coupling constant, but if RDCs larger than ±30 Hz are anticipated and/or more accurate J/D values are required, then multiple experiments with different settings of the BIRD/INEPT delays should be performed.
Finally, the impact of miscalibration of 1H and 13C pulses on the accuracy of the measured J values was studied by setting the corresponding pulses longer of ca. 5% in separate experiments, varying the 1H offset in 40 steps over an 80 Hz range. It was found that at 5% misset, the value of 1JCH remained basically unaffected, yielding Jav = 144.01 Hz with maximum deviation of 0.90 Hz and standard deviation σJ = 0.37 Hz for a 5% error in 1H pulse width, and Jav = 143.96 Hz with maximum deviation of 1.15 Hz and standard deviation σJ = 0.57 Hz for a similar error in 13C pulse width.
Full details of the phase cycling and sequencing combinations tested, together with the averages and standard deviations of 1JCH values and chemical shifts (δ) obtained for the CH3-group of methyl α-D-mannopyranoside in the proton offset-dependence measurements, are given in Table S1 of the ESI.† The best phase sequence was based on MLEV-8, in a minor extension of the sequence used in ref. 14. This exploits the cyclicity of the MLEV scheme to minimize accumulation of errors, the error in the first pair of J-refocusing elements being partially cancelled by the second, and the remaining error partially cancelled by the second block of four pulses.
Having established the robustness of the new experiments, as a real case study, the performance and reproducibility of the optimized real-time BIRD(d) CLIP/CLAP-HSQC sequences of Fig. 1 were investigated on a 55-residue 15N-labeled protein (PAF)29,30 sample in H2O by running multiple measurements. Experiments for all three 1H–15N CLIP/CLAP-HSQC variants, conventional proton-coupled, BIRD(d) interferogram-based, and real-time homonuclear decoupled CLIP/CLAP-HSQCs, were performed for comparison. In general, triplicate measurements were carried out to estimate the uncertainties of the 1JNH values obtained. 1JNH values determined from a BIRD(d) CLIP-HSQC spectrum recorded with interferogram-based acquisition, where cumulative errors are not a problem, were used as reference values. It has been recently shown3 that the latter experiment provides reliable and precise 1JXH values by direct measurement of the separations of peak maxima in the X-coupled doublets. In the present study, for fast and automated evaluation of coupling constants the peak positions were simply determined using automatic peak picking in all protein spectra. In the case of the conventional, proton-coupled experiment, spectra were processed with sufficient line broadening in F2 to ensure that proton–proton couplings were not resolved.
To demonstrate the spectral simplification and resolution enhancement achieved by the proposed method, an overlay of the proton-coupled (black) and real-time broadband proton-homodecoupled (red) 1H–15N CLIP-HSQC spectra of 15N-labeled PAF, together with selected expansions and doublets, is shown in Fig. 2. For detailed comparison of the three CLIP/CLAP-HSQC variants, representative CLIP doublets are also shown in Fig. 3.
 | ||
| Fig. 2 Overlay of 1H–15N CLIP-HSQC spectra of 15N-labeled penicillium antifungal protein (PAF, 1.7 mM in 95% H2O/5% D2O using 20 mM Na3PO4 pH 6.0 buffer, 40 mM NaCl, 0.04% NaN3, as 275 μl of solution in Shigemi NMR tube), acquired by the conventional proton-coupled (black, lower) and real-time broadband proton-homodecoupled (red, upper) pulse sequence. The decoupled spectrum is shifted in the nitrogen dimension for better visualization. An expansion and representative doublets are shown to demonstrate the simplification of peak patterns achieved by broadband proton homodecoupling using the pulse sequence of Fig. 1. The following parameters were used in these experiments: spectral width in 1H (15N) dimension = 4.7880 (21.0) ppm, number of complex data points in 1H dimension = 2048, number of t1 increments = 256, number of scans = 8, relaxation delay = 1.7 s. Data acquisition in the pure shift experiment was divided into 16 chunks and the duration of each chunk was 26.7 ms. | ||
 | ||
| Fig. 3 Representative doublets of Phe-25 (A) and Val-52 (B) in PAF extracted from the conventional proton-coupled1 (blue and red), interferogram-based decoupled3 (black) and real-time decoupled (green) 1H–15N CLIP-HSQC spectra of 15N-labeled protein. Relevant experimental parameters are the same as given for Fig. 2. In the interferogram-based experiment 4 scans and 16 chunks were used for construction of the FIDs. In the case of the proton-coupled variant, a strong Gaussian apodization function was applied during processing (red) to obscure splitting due to proton–proton coupling to aid automated spectral analysis. | ||
The almost perfect match between pure shift doublets measured in real-time (green) and in interferogram (black) experiments is remarkable. In contrast, the proton-coupled doublets (blue in Fig. 3) show doublet fine structure due to proton–proton coupling that is slightly asymmetric, possibly as a result of relaxation effects or of homonuclear J modulation during the INEPT sequence elements. Note that the latter could be minimized by using perfect-echo INEPT sequences in the heteronuclear transfer periods, as proposed recently.31–33 Strong proton–proton coupling in the 13C isotopomer34,35 is a further source of complication, best dealt with by numerical simulation.
When severe weighting is used (red) to broaden out the homonuclear coupling structure to allow automated peak-picking, a systematic underestimation of 1JCH is evident. Consequently, the coupling constants extracted from the regular, proton-coupled CLIP/CLAP-HSQC spectra with automated peak analysis are biased (specifically, slightly underestimated), as demonstrated in Fig. 4, where the 1JNH values of the well resolved resonances of PAF determined by automated analysis of the three CLIP/CLAP-HSQC variants are shown as a function of residue number. Note that almost all the 1JNH values determined from the proton-coupled spectra (labeled with black diamonds in Fig. 4) are slightly smaller – with a maximum deviation of 1.6 Hz, and deviations >1 Hz for 13 residues out of 47 – than the 1JNH values measured from the decoupled spectra. Noticeably smaller deviations, with a maximum of 0.46 Hz and only 4 residues >0.35 Hz, were found between corresponding 1JNH values extracted from the real-time and the interferogram proton-homodecoupled spectra. The average standard deviation for the proton-homodecoupled experiments, based on six measurements (three real-time BIRD(d) CLIP-HSQCs and three real-time BIRD(d) CLAP-HSQCs), was about half of that for the proton-coupled experiments. All the relevant data are summarized for comparison in Table S4 of the ESI.†
 | ||
| Fig. 4 Comparison of 1JNH values measured using the three types of 1H–15N CLIP/CLAP-HSQC spectra of 15N-labeled PAF (1.7 mM in 95% H2O/5% D2O) as a function of residue number. Mean values of 6 experiments in the case of coupled (black diamonds) and real-time decoupled (red squares), and mean values of 3 experiments in the case of interferogram-based decoupled (blue triangles) method are plotted. Standard deviations based on multiple measurements are depicted as error bars. Peak positions were determined using automated peak-picking in each case, and a script (named RDC-calculator) implemented in CCPNMR was used for the calculation of coupling constants. 1JNH values are not shown in some cases due to low intensity signals/line broadening (Lys-2, Ser-10, Lys-11, Asp-32) or signal overlap (Tyr-3, Thr-24). | ||
To mimic the variation in coupling constant typically expected in proteins under anisotropic conditions, multiple experiments were performed with different BIRD/INEPT delays, corresponding to the range of couplings from 66 Hz to 116 Hz in steps of 5 Hz. The 1JNH values measured for BIRD/INEPT delays set for 86–111 Hz for residues of PAF are plotted in Fig. 5; the maximum average absolute deviation from the mean of 1JNH is only 0.33 Hz over this range. Importantly, even over the 50 Hz range of 66–116 Hz the maximum deviation increases only slightly, up to 1.05 Hz. All the data are summarized in Table S5 of the ESI.† These data, with only modest errors in 1JNH values, thus provide good evidence for the applicability of the proposed real-time experiments to N–H RDC measurements in proteins.
 | ||
| Fig. 5 Comparison of 1JNH values measured for well-resolved resonances of 15N-labeled PAF (1.7 mM in 95% H2O/5% D2O) using the real-time broadband proton-homodecoupled CLIP- and CLAP-HSQC sequences of Fig. 1 with different settings of the INEPT/BIRD delays. The delays (τ, τ′, 2τ in Fig. 1) were varied to correspond to couplings from 86 to 111 Hz in steps of 5 Hz. 1JNH values are not shown in some cases due to low intensity signals/line broadening (Lys-2, Ser-10, Lys-11, Asp-32) or signal overlap (Tyr-3, Thr-24). | ||
As a proof of principle, real-time BIRD(d) CLIP/CLAP-HSQC experiments were performed with the pulse sequence of Fig. 1 on a 15N-PAF sample prepared in the weakly orienting medium36 containing 5% C8E5 and octanol in H2O. Representative excerpts from the 15N-CLIP-HSQC spectra – proton-coupled in black and real-time BIRD(d) homodecoupled in red – together with selected doublets are shown in Fig. 6. The sequence variation of 1TNH and 1JNH, and their differences 1DNH as a measure of the RDCs, are plotted in Fig. 7 for those residues that are not broadened and/or overlapping. As expected the average standard deviation of 1TNH-values obtained in the anisotropic medium was somewhat larger (1.00 Hz) than that obtained under isotropic condition (0.17 Hz). All relevant data, including standard deviations (RMSD) assessed from multiple experiments, are supplied in Table S6 of the ESI,† and provide further evidence that the proposed real-time BIRD(d) CLIP/CLAP-HSQC experiments are suitable for reliable automated one-bond 1H–15N RDC measurements in proteins. Where necessary to avoid resonance overlap due to the increased number of cross-peaks in coupled spectra, the IPAP-approach37 (taking the sum and difference of CLIP- and CLAP-HSQC spectra) can be utilized. Alternatively, variants of F1-coupled HSQC2,38,39 or J-resolved40–42 spectra may also offer solutions to the resonance overlap problem.
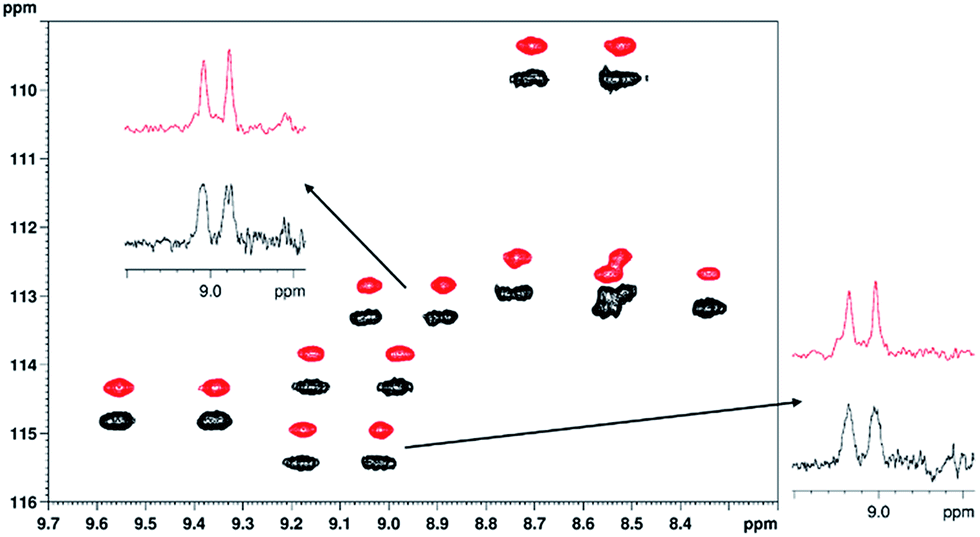 | ||
| Fig. 6 Overlay of parts of the 1H–15N CLIP-HSQC spectra of 15N-labeled PAF protein (3.2 mg) dissolved in weakly orienting medium 5% C8E5 and octanol in H2O/D2O (total 500 μl, pH = 6, phosphate buffer),36 acquired using the conventional proton-coupled (black, lower) and real-time broadband proton-homodecoupled (red, upper) sequence. The decoupled spectrum is shifted in the nitrogen dimension for better visualization. Representative doublets are shown to demonstrate the simplification of peak patterns achieved by broadband proton homodecoupling using the pulse sequence of Fig. 1. The following parameters were used in these experiments: spectral width in 1H (15N) dimension = 4.7880 (21.0) ppm, number of complex data points in 1H dimension = 1024 (2048 for coupled), number of t1 increments = 256, number of scans = 32, relaxation delay = 1.7 s. Data acquisition in the pure shift experiment was divided into 8 chunks and the duration of each chunk was 26.7 ms. Traces are extracted at δ15N = 113.3 ppm (top) and 115.4 ppm (right), respectively. | ||
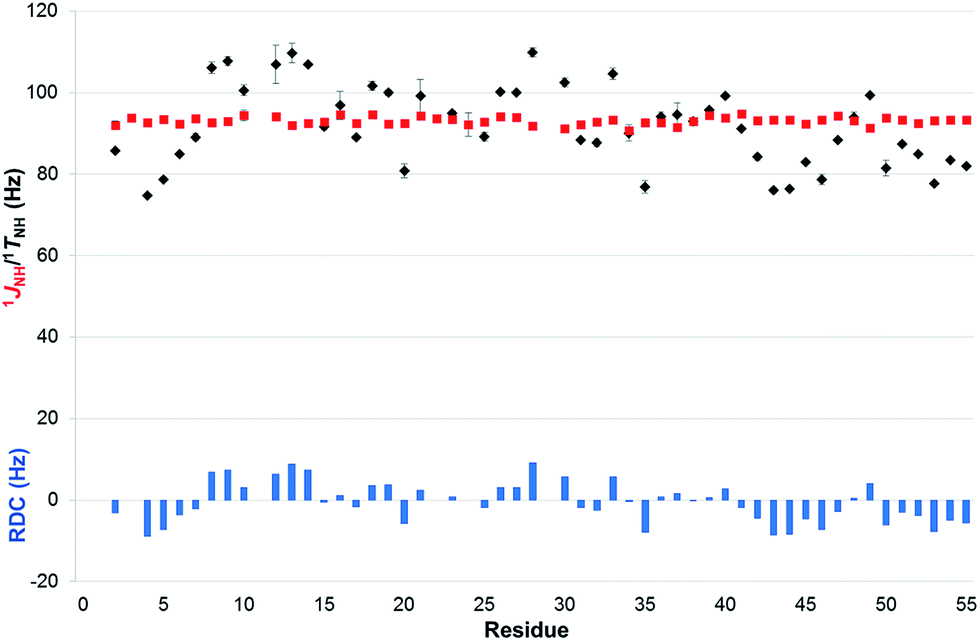 | ||
| Fig. 7 Comparison of 1JNH (red squares) and 1TNH = (1JNH + 2 × 1DNH) values (black diamonds) measured with the real-time broadband proton-homodecoupled CLIP- and CLAP-HSQC sequences proposed in 15N-labeled PAF under isotropic (95% H2O/5% D2O) and anisotropic conditions (5% C8E5, octanol),36 respectively. Error bars shown are estimated from data scattering in multiple measurements. RDCs (blue columns), representing the value of 1DNH, are also shown as a function of residue number. | ||
Having established that real-time BIRD(d) CLIP/CLAP-HSQC experiments are useful for 1JNH and 1DNH extraction for weakly aligned proteins, we now turn to the case of 1JCH and 1DCH measurements in a small organic molecule without isotopic enrichment. The challenges associated with one-bond coupling measurement in small organic molecules are different from those arising for backbone amides in proteins, as greater robustness of the experiment with respect to differences in coupling network and one-bond coupling is required in this case. On the other hand, the effects of transverse relaxation are much less pronounced in small molecules, increasing the benefits of homodecoupling.
To evaluate the techniques described, we measured real-time BIRD(d) CLIP/CLAP-HSQC spectra for the same samples used in a previous study,5 containing (+)-isopinocampheol dissolved in dichloromethane-d2 for measurements in isotropic phase, and in a high molecular weight poly-γ-benzyl-D-glutamate/dichloromethane-d2 liquid crystalline medium43,44 for measurement under weak alignment. Initial concerns about sample stability arose when a slightly different deuterium quadrupolar splitting ΔνQ = 110.5 Hz, instead of ΔνQ = 107.6 Hz, was observed compared to the previous measurement 20 months earlier. The difference seen can probably be explained by a slight change (∼0.5 K) that was found in the temperature calibration of the setup used, although ageing of the sample cannot be excluded even though the sample was thoroughly degassed and sealed. For comparison purposes, the total and dipolar couplings reproduced from the previous study have therefore been scaled here by the ratio of the quadrupolar splittings observed, to take into account the slight increase in order parameter.
For the isotropic sample, a reduction of the spectral quality obtained in the real-time BIRD(d) decoupled experiments is seen compared to the experiments with interferogram-based BIRD(d) decoupling. Representative traces for direct comparison are given in Fig. S3.† The 1JCH coupling constants which are extracted from real-time BIRD(d) CLIP/CLAP-HSQCs reflect this, showing marked deviations from the values obtained from the spectra with interferogram-based homodecoupling, which in turn agree much better with the experiments without homodecoupling (real-time BIRD(d) CLIP/CLAP-HSQC: maximum deviations of 0.9 Hz and standard deviations σJ of 0.4 Hz; CLIP/CLAP-HSQC without homodecoupling: maximum deviations of 0.2 Hz and standard deviations σJ: of 0.1 Hz; see columns 1, 4 and 5 of Table S7†). Thus for small organic molecules, the compromise between experiment duration and experimental accuracy required when moving from interferogram to real-time BIRD(d) decoupling is much less favorable than it is for proteins.
In addition to comparing different experiment types, Table S7† offers two further comparisons. First, it shows that for real-time decoupling, longer data chunks provide more accurate coupling constant measurement, since, as noted above, extending the period of the decoupling cycle reduces the amplitudes of the unwanted extra irradiation frequencies. The upper limit on chunk duration is determined by the acceptable level of the chunking sidebands that are caused by coupling evolution during data chunks. As a consequence, reducing the acquisition chunk duration from 20.48 ms to 15.36 ms and further to 10.24 ms leads to a progressive reduction in coupling constant measurement accuracy (compare columns 5–7 of Table S7†). Changing the chunk duration between scans, as proposed recently,45 can be also useful in reducing decoupling sidebands. Second, it can be seen that using an 8 step phase cycle in place of the 16 step cycling used in the rest of this study leads to virtually identical results (compare columns 5 and 8 of Table S7†).
In the case of the weakly aligned sample it is reassuring to see that the real-time BIRD(d) approach provides a significant linewidth reduction as compared to the experiment without homodecoupling, without any requirement to increase the experiment duration through the introduction of an additional homodecoupling time dimension (as in interferogram experiments). Fig. 8 illustrates that for weakly aligned samples, just as for the PAF protein, marked reductions in signal widths are possible with real-time BIRD(d) decoupling. Decoupling sidebands are mostly below 5% of the main signal intensity, and therefore are likely to be masked by spectral noise for low concentration samples.
 | ||
| Fig. 8 CLAP-HSQC spectra with real-time BIRD decoupling (pos. blue, neg. magenta; 20.48 ms chunk duration) and without homodecoupling (pos. black, neg. cyan) in the proton dimension. The spectra were acquired for a sample containing (+)-isopinocampheol dissolved in a PBLG/CD2Cl2 lyotropic liquid crystalline medium (9.05% wt, ΔνQ = 110.5 Hz). The selected F2 traces shown are plotted at the same absolute intensity scale. In the proton numbering given, the descriptors “s” and “a” refer to syn or anti with respect to the dimethylmethylene bridge. | ||
As in our previous study,5 in anisotropic solution, the most challenging protons for homodecoupling techniques are protons H7a and H7s (for numbering, see Fig. 8), which show a geminal splitting of 2TCH ≈ −38.4 Hz under the current alignment conditions. The poor lineshape observed, which is illustrated as an insert in Fig. 8, prompted us to estimate very conservative experimental errors for the one-bond couplings extracted for these protons (5 to 7 Hz for the data sets acquired with 20.48 ms chunk length), while the error estimates are much smaller for other signals. In general, the BIRD homodecoupling is not able to suppress geminal couplings. From signals such as H4a/s and H8, it can be seen however, that for smaller geminal couplings the residual homonuclear splittings do not prevent 1TCH extraction.
While for 1JCH couplings it was found that the measurement accuracy was reduced compared to the experiment without homodecoupling, this was not the case for the 1TCH couplings listed in Table S8.† Restricting the discussion to the results obtained with a chunk length of 20.48 ms, data collected with real-time BIRD(d) decoupling and data collected with no homodecoupling agree equally well with the data from the perfect BIRD CLIP-HSQC experiment chosen as reference (real-time BIRD(d) CLIP/CLAP-HSQC: maximum deviations of 1.5 to 2.0 Hz and standard deviations σJ of 0.7 to 0.9 Hz; CLIP/CLAP-HSQC without homodecoupling: maximum deviations of 1.0 to 3.4 Hz and standard deviations σJ of 0.5 to 1.5 Hz; see columns 1, 4, 5 and 8 of Table S8†). Taking the same experiment time, for small molecules the real-time decoupled experiment thus seems to perform as well as the experiment without decoupling, even in the absence of signal overlap.
The RDCs listed in Table S9† finally show that the real-time BIRD(d) decoupled experiment with a chunk length of 20.48 ms provides one-bond RDCs which agree sufficiently well with the reference data (maximum deviations of 1.1 to 1.2 Hz and standard deviations σJ of 0.5 to 0.6 Hz; see columns 5 and 8 of Table S9†) that, the use of this experiment in structural studies of small molecules is practical. Based on these data we suggest using a minimum confidence interval of ±1 Hz for RDCs measured in small molecules using real-time BIRD(d) homodecoupling. Compared to the results obtained for backbone N–H RDC measurement in the test protein system this is a slight reduction in accuracy, which can be attributed to the differences in complexity of spin system between these two test samples. As in the case of the protein study, if larger variations in resonance offset or in one-bond coupling than used in this study occur, we recommend either we recommend either that an independent evaluation of the experiment accuracy be performed, or that several experiments with different experimental parameters be used (the maximum offsets used here were 1540 Hz for 1H and 4000 Hz for 13C, and one-bond total couplings varied between 106 and 161 Hz).
In addition to comparing the one-bond couplings measured with those from a high accuracy reference dataset, we chose to evaluate the one-bond RDC measurement accuracy by testing the consistency of the different datasets against a structural model already used in previous studies.5,43,44,46 The agreement of the experimentally determined 1DCH values with the structural model, as expressed by the RMSD between the experimental and the back-calculated RDCs, confirms that the data measured using real-time BIRD(d) decoupled CLIP-HSQCs agree with the structure as well as the data obtained from CLIP-HSQCs without homodecoupling. The RMSD ranges from 0.15 Hz to 0.28 Hz for real-time BIRD CLIP/CLAP-HSQC experiments with 20.48 ms chunk length, and from 0.16 Hz to 0.26 Hz for CLIP/CLAP-HSQC experiments without homodecoupling (see columns 1, 7–10, 13 and 14 of Table S10†).
This sample study of (+)-isopinocampheol illustrates that real-time BIRD(d) CLIP/CLAP-HSQC experiments can provide one-bond RDCs with sufficient accuracy for small molecule structural studies. While RDC measurement accuracies are higher in experiments using interferogram-based homodecoupling, the accuracy in the real-time BIRD(d)-decoupled experiment can be comparable to that obtained in experiments without homodecoupling. The technique presented thus paves the way for rapid and pure shift RDC measurements in small molecules. Full experimental data files can be freely downloaded from DOIs 10.15127/1.303524 and 10.15127/1.303527.
Conclusions
We have described experimental methods for obtaining pure shift CLIP/CLAP-HSQC spectra utilizing isotope-selective BIRD(d)-based J-refocusing elements for real-time broadband proton–proton decoupling, leading to significant resolution enhancement in the directly detected (1H) dimension at no extra cost in measurement time. The resultant real-time BIRD(d) CLIP/CLAP-HSQC spectra provide broadband proton-homodecoupled pure in- or anti-phase doublets with improved resolution, allowing direct and relatively precise measurement of one-bond heteronuclear couplings using quick automatic peak picking algorithms during spectral analysis of protein N–H regions.The utility of the real-time pure shift sequences presented here has been demonstrated for coupling constant measurements under both isotropic and anisotropic conditions, on a protein sample and on a small test molecule. The numerous tests and comparisons with reference data demonstrate that the proposed real-time BIRD(d) CLIP/CLAP-HSQC experiments show good precision, reproducibility and robustness for the measurement of coupling constants. Applicable to samples in both isotropic and anisotropic media, they provide reliable and precise data for probing the structures and dynamics of molecules via RDC analysis.
Acknowledgements
This research was supported by the National Research, Development and Innovation Office of Hungary (grant numbers NKFI/OTKA NN 109671 and K 105459 to K. E. K.), by the Richter Gedeon Talentum Alapítvány (Ph.D. scholarship to I. T. and M. H. R.), by the Engineering and Physical Sciences Research Council (grant numbers EP/I007989/1 and EP/L018500/1 to G. A. M.), by the ERC (grant number 257041 to C. M. T.), and by the Merck Foundation for Arts and Science (Ph.D. scholarship to L. K.). The authors thank Dr Florentine Marx for providing the 15N-labeled PAF protein sample.Notes and references
- A. Enthart, J. C. Freudenberger, J. Furrer, H. Kessler and B. Luy, J. Magn. Reson., 2008, 192, 314–322 CrossRef CAS PubMed
.
-
L. Castañar and T. Parella, in Annual Reports on NMR Spectroscopy, ed. A. W. Graham, Academic Press, 2015, vol. 84, pp. 163–232 Search PubMed
- I. Timári, L. Kaltschnee, A. Kolmer, R. W. Adams, M. Nilsson, C. M. Thiele, G. A. Morris and K. E. Kövér, J. Magn. Reson., 2014, 239, 130–138 CrossRef PubMed
- T. Reinsperger and B. Luy, J. Magn. Reson., 2014, 239, 110–120 CrossRef CAS PubMed
- L. Kaltschnee, A. Kolmer, I. Timári, V. Schmidts, R. W. Adams, M. Nilsson, K. E. Kövér, G. A. Morris and C. M. Thiele, Chem. Commun., 2014, 50, 15702–15705 RSC
- M. Nilsson and G. A. Morris, Chem. Commun., 2007, 933–935 RSC
- J. A. Aguilar, S. Faulkner, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2010, 49, 3901–3903 CrossRef CAS PubMed
- A. Lupulescu, G. L. Olsen and L. Frydman, J. Magn. Reson., 2012, 218, 141–146 CrossRef CAS PubMed
- L. Paudel, R. W. Adams, P. Király, J. A. Aguilar, M. Foroozandeh, M. J. Cliff, M. Nilsson, P. Sándor, J. P. Waltho and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 11616–11619 CrossRef CAS PubMed
- J. R. Garbow, D. P. Weitekamp and A. Pines, Chem. Phys. Lett., 1982, 93, 504–509 CrossRef CAS
- K. Zangger and H. Sterk, J. Magn. Reson., 1997, 124, 486–489 CrossRef CAS
- L. Castañar, P. Nolis, A. Virgili and T. Parella, Chem.–Eur. J., 2013, 19, 17283–17286 CrossRef PubMed
- R. W. Adams, L. Byrne, P. Király, M. Foroozandeh, L. Paudel, M. Nilsson, J. Clayden and G. A. Morris, Chem. Commun., 2014, 50, 2512–2514 RSC
- J. Ying, J. Roche and A. Bax, J. Magn. Reson., 2014, 241, 97–102 CrossRef CAS PubMed
- N. H. Meyer and K. Zangger, Angew. Chem., Int. Ed., 2013, 52, 7143–7146 CrossRef CAS PubMed
-
R. W. Adams, in eMagRes, John Wiley & Sons, Ltd, 2014, vol. 3, pp. 295–310 Search PubMed
- L. Castañar and T. Parella, Magn. Reson. Chem., 2015, 53, 399–426 CrossRef PubMed
- K. Zangger, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 86–87, 1–20 CrossRef CAS PubMed
- P. Kiraly, R. W. Adams, L. Paudel, M. Foroozandeh, J. A. Aguilar, I. Timári, M. J. Cliff, M. Nilsson, P. Sándor, G. Batta, J. P. Waltho, K. E. Kövér and G. A. Morris, J. Biomol. NMR, 2015, 62, 43–52 CrossRef CAS PubMed
- M. Perez-Trujillo, L. Castañar, E. Monteagudo, L. T. Kuhn, P. Nolis, A. Virgili, R. T. Williamson and T. Parella, Chem. Commun., 2014, 50, 10214–10217 RSC
- D. Uhrín, T. Liptaj and K. E. Kövér, J. Magn. Reson., Ser. A, 1993, 101, 41–46 CrossRef
- G. A. Morris, G. L. Nayler, A. J. Shaka, J. Keeler and R. Freeman, J. Magn. Reson., 1984, 58, 155–160 CAS
- A. J. Shaka, J. Keeler, R. Freeman, G. A. Morris and G. L. Nayler, J. Magn. Reson., 1984, 58, 161–166 CAS
- V. M. Kakita and J. Bharatam, Magn. Reson. Chem., 2014, 52, 389–394 CrossRef CAS PubMed
- S. Glanzer and K. Zangger, J. Am. Chem. Soc., 2015, 137, 5163–5169 CrossRef CAS PubMed
- S. Glanzer and K. Zangger, ChemPhysChem, 2015, 16, 3313–3317 CrossRef CAS PubMed
- S. Glanzer, O. Kunert and K. Zangger, J. Magn. Reson., 2016, 268, 88–94 CrossRef CAS PubMed
- G. A. Morris and R. Freeman, J. Magn. Reson., 1978, 29, 433–462 CAS
- G. Batta, T. Barna, Z. Gáspári, S. Sándor, K. E. Kövér, U. Binder, B. Sarg, L. Kaiserer, A. K. Chhillar, A. Eigentler, É. Leiter, N. Hegedüs, I. Pócsi, H. Lindner and F. Marx, FEBS J., 2009, 276, 2875–2890 CrossRef CAS PubMed
- G. Váradi, G. K. Tóth, Z. Kele, L. Galgóczy, Á. Fizil and G. Batta, Chem.–Eur. J., 2013, 19, 12684–12692 CrossRef PubMed
- B. Baishya and C. L. Khetrapal, J. Magn. Reson., 2014, 242, 143–154 CrossRef CAS PubMed
- L. Castañar, E. Sistaré, A. Virgili, R. T. Williamson and T. Parella, Magn. Reson. Chem., 2015, 53, 115–119 CrossRef PubMed
- C. Mauve, S. Khlifi, F. Gilard, G. Mouille and J. Farjon, Chem. Commun., 2016, 52, 6142–6145 RSC
- G. A. Morris and K. I. Smith, Mol. Phys., 1987, 61, 467–483 CrossRef CAS
- B. Yu, H. van Ingen, S. Vivekanandan, C. Rademacher, S. E. Norris and D. I. Freedberg, J. Magn. Reson., 2012, 215, 10–22 CrossRef CAS PubMed
- M. Rückert and G. Otting, J. Am. Chem. Soc., 2000, 122, 7793–7797 CrossRef
- M. Ottiger, F. Delaglio and A. Bax, J. Magn. Reson., 1998, 131, 373–378 CrossRef CAS PubMed
- K. Fehér, S. Berger and K. E. Kövér, J. Magn. Reson., 2003, 163, 340–346 CrossRef
- C. M. Thiele and W. Bermel, J. Magn. Reson., 2012, 216, 134–143 CrossRef CAS PubMed
- K. E. Kövér and K. Fehér, J. Magn. Reson., 2004, 168, 307–313 CrossRef PubMed
- J. Furrer, M. John, H. Kessler and B. Luy, J. Biomol. NMR, 2007, 37, 231–243 CrossRef CAS PubMed
- J. D. Snider, E. Troche-Pesqueira, S. R. Woodruff, C. Gayathri, N. V. Tsarevsky and R. R. Gil, Magn. Reson. Chem., 2012, 50, S86–S91 CrossRef CAS PubMed
- A. Marx, V. Schmidts and C. M. Thiele, Magn. Reson. Chem., 2009, 47, 734–740 CrossRef CAS PubMed
- A. Marx and C. Thiele, Chem.–Eur. J., 2009, 15, 254–260 CrossRef CAS PubMed
- J. Mauhart, S. Glanzer, P. Sakhaii, W. Bermel and K. Zangger, J. Magn. Reson., 2015, 259, 207–215 CrossRef CAS PubMed
- N. C. Meyer, A. Krupp, V. Schmidts, C. M. Thiele and M. Reggelin, Angew. Chem., Int. Ed., 2012, 51, 8334–8338 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental parameters, phase programs tested, tables of one-bond heteronuclear coupling constants measured, Bruker pulse sequence codes. See DOI: 10.1039/c6ra14329f |
| This journal is © The Royal Society of Chemistry 2016 |