
A novel technique in the foaming process of EPDM/PP via microwave radiation: the effect of blend compatibilization and additive encapsulation
 c
and
c
and
Abstract
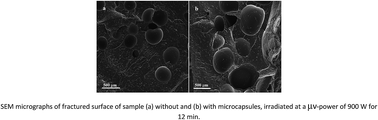
Uniform voids, greater cell sizes, and their better distribution in the foaming of EPDM/PP could be achieved using microwave radiation with the peerless approach of employing encapsulated additives. The introduction of PP-g-MA as a compatibilizer proved to be an effective way to achieve better dispersion of PP droplets in the matrix of EPDM, and also to improve the elongational viscosity at higher extension rates. The ultimate foam density was lowered from 0.71 to about 0.59 g cm−3 as a result of applying foaming agents, along with iron oxide particles, encapsulated by urea–formaldehyde polymer at a microwave oven power of 900 W. It is of vital significance that the mean cell sizes from this technique reached 435 microns, compared to 277 microns attained in conventional methods.
 Please wait while we load your content...
Please wait while we load your content...

