
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cofactor-specific covalent anchoring of cytochrome b562 on a single-walled carbon nanotube by click chemistry†
Akira Onoda*a,
Nozomu Inouea,
Stéphane Campidellib and
Takashi Hayashi*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, 565-0871, Japan. E-mail: onoda@chem.eng.osaka-u.ac.jp; thayashi@chem.eng.osaka-u.ac.jp
bLICSEN, NIMBE, CEA, CNRS, Université Paris-Saclay, CEA Saclay, 91191 Gif-sur-Yvette Cedex, France
First published on 4th July 2016
Abstract
Redox-active cytochrome b562 with a tethered azide group on the heme propionate side chain is covalently linked to an acetylene moiety introduced on the sidewall of a single-walled carbon nanotube (SWNT) by copper-catalyzed click chemistry forming a triazole ring with the heme active site directly linked to the SWNT. The cytochrome b562–SWNT hybrid is characterized by electrochemistry and atomic force microscopy.
Interfacing redox-active enzymes with electrode materials is a key technology used in the development of high performance biosensors and biofuel cells.1–3 Recent advances in carbon nanomaterials have enabled us to design hybrid systems with linked enzymes. Carbon nanotubes (CNTs), of the single- or multi-walled type, are promising building blocks for fabrication of hybrid materials.4–8 CNTs have large surface areas, high strength, chemical stability, and attractive electronic properties.
CNTs have also provided a wide variety of synthetic tools applicable for introduction of a range of substituents for linking the enzymes. A copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction is a widely utilized method used to form a covalent linkage which includes a triazole ring between building blocks containing azide and alkyne groups.9,10 The CuAAC reaction has thus been used in organic synthesis, bioconjugation chemistry, and surface chemistry. This powerful coupling reaction can be applied in efforts to efficiently tailor the chemical modification of single-walled CNTs (SWNTs) to construct hybrid materials11–13 including enzymes.14
Redox-active hemoproteins form a major class of enzymes that are useful for constructing enzyme-immobilized electrodes due to their diverse functions including electron transfer, catalysis, and sensing.15–25 Many hemoproteins possess a replaceable heme b cofactor in the heme pocket, enabling immobilization on the electrode via the heme–heme pocket interaction.26–45 In this paper, we demonstrate specifically oriented covalent immobilization of azide-linked cytochrome b562 (CYT) on the sidewall of SWNT using the CuAAC reaction (Fig. 1). The advantage of this method which uses a replaceable heme tethered to an azide moiety, lies in the wide range of applications for functionalization of wild-type hemoproteins. The characterization and electrochemical properties of the covalently-linked hybrid materials of SWNT and cytochrome b562 are described.
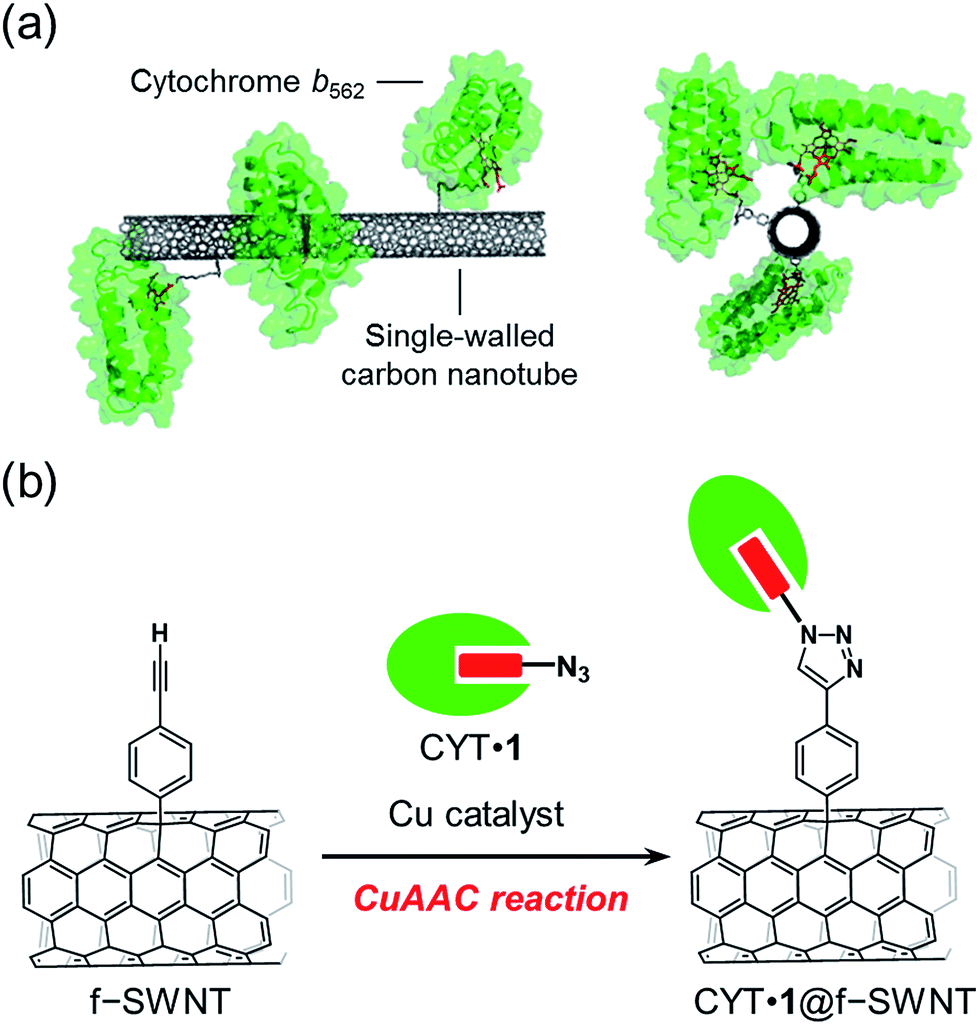 | ||
| Fig. 1 (a) SWNT with covalently-linked cytochrome b562 and (b) the preparation scheme using a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. | ||
Arc discharge SWNT was purified as described previously.11 They consist of a mixture of metallic and semiconducting nanotubes with a narrow diameter distribution peaked at 1.4 nm. SWNT was functionalized in a reaction with 4-((2-trimethylsilyl)ethynyl)benzenediazonium prepared in situ by mixing 4-((2-trimethylsilyl)ethynyl)aniline and isoamyl nitrite.11 The trimethylsilyl group of the modified SWNT was deprotected to yield functionalized SWNT tethering 4-ethynylphenyl groups (f-SWNT). The covalent grafting of 4-ethynylphenyl groups onto the SWNT sidewalls was confirmed by observing a decrease in intensity of the absorption peaks at 730, 1030, and 1090 nm for SWNT and an increase in intensity of the D-band in a Raman spectrum (Fig. 2 and S1, ESI†). Thermogravimetric analysis (TGA) between 100 and 600 °C indicates that the weight loss corresponding to the functionalization of 4-ethynylphenyl group in f-SWNT is about 13%, suggesting that approximately one ethynylphenyl group is attached per 100 carbon atoms of the SWNT sidewall (Fig. S1, ESI†).
 | ||
| Fig. 2 Raman spectra with excitation at 532 nm of purified SWNT (black) and f-SWNT (red). The increase of the D-band at 1345 cm−1 for f-SWNT is derived from the covalent functionalization with the phenylacetylene diazonium derivative. | ||
Azide-linked protoporphyrin IX was synthesized according to the previously reported method with a slight modification.46 The corresponding propionate-modified porphyrin was treated with FeCl2. After purification, the final product 1 was obtained. The product was characterized by ESI-TOF MS (calcd 817.29; found 817.29). The characteristic absorptions at 409, 538, and 553 nm in pyridine confirm the insertion of iron to generate the pyridine-bound ferric (Fe3+) heme (Fig. S2, ESI†).
Heme 1 linked to the azide group was inserted into apocytochrome b562 (apoCYT) according to the conventional method,47 followed by purification using a gel filtration column to produce the reconstituted cyt b562, which is designated CYT·1 (Fig. 3a). The reconstituted protein was isolated as the ferric (Fe3+) form with axial ligation by His and Met, as confirmed by the standard absorption peaks at 418, 530, and 563 nm. This indicates that heme is located in the normal position of the heme pocket of cyt b562 (Fig. 3b). Insertion of the modified heme was also determined by ESI-TOF MS experiments for CYT·1 (calcd for C550H883N154O175S3 [M + 7 H+]7+: 1800.62; found: 1800.48) (Fig. 3c). These reconstituted proteins were found to be stable for several days at 4 °C.
 | ||
| Fig. 3 (a) Preparation of CYT·1. (b) UV-vis spectra of CYT·1. (c) ESI-TOF MS of CYT and CYT·1. | ||
CYT·1 with the tethered azide moiety was covalently introduced onto the phenylacetylene-modified SWNT in the CuAAC reaction performed in a buffer solution. Sodium dodecyl sulfate surfactant, which is commonly used as a solubilizer of SWNT, is known to denature proteins even at low concentrations. Thus, to increase the solubility, f-SWNT was dispersed in a buffer containing 2 wt% sodium cholate that wraps SWNT in water. As a result, it was confirmed that CYT is stably folded over three days in a buffer containing 2 wt% sodium cholate by monitoring the Soret and Q bands (Fig. S3, ESI†). Before the reaction with SWNT, the CuAAC reaction of CYT·1 with 5-hexynoic acid in the presence of 2 wt% sodium cholate, tris(3-hydroxypropyltriazolylmethyl)amine, and sodium ascorbate as a reductant was tested at 4 °C for 48 h. The successful coupling with a triazole moiety was confirmed by EST-TOF MS (Fig. S4, ESI†). We then performed the CuAAC reaction with f-SWNT to connect SWNT to the propionate group of the heme in CYT via a covalent linkage including a triazole moiety. The f-SWNT modified with CYT was purified by dialysis to remove excess CuAAC reagents.
The conjugation by CuAAC of CYT·1 and f-SWNT was confirmed using by differential pulse voltammetry (DPV). f-SWNT reacted in the absence of CuSO4 was prepared as a reference (Fig. 4). The CYT·1@f-SWNT conjugate was suspended in 50% Nafion® in MOPS buffer and the suspension was immersed on a glassy carbon electrode for the electrochemical measurements. CYT·1@f-SWNT, in which CYT is covalently-linked with SWNT, exhibits redox peaks at −0.4 V vs. Ag/AgCl both in positive and negative scans. The redox peaks are assigned to the redox couple of the heme. In contrast, the sample prepared without CuSO4 does not have similar redox peaks. These results clearly indicate that the strategy for combining SWNT and CYT in the CuAAC reaction provides stable hybrids which are linked by a triazole moiety.
 | ||
| Fig. 4 DPVs of (a) CYT·1@f-SWNT and (b) f-SWNT. Scan rate: 1 to 10 V s−1 in a degassed 100 mM KPi buffer (pH 7.0) at 25 °C. WE: sample + Nafion®/GC, CE: Pt mesh, RE: Ag|AgCl (3 M NaCl), 50 mM MOPS (pH 7.0), scan rate: 10 mV s−1. | ||
The SWNT–CYT conjugate was directly analyzed by atomic force microscopy (AFM) to characterize its morphology (Fig. 5). The samples were deposited on a mica surface and the non-covalently adsorbed proteins were removed by washing with deionized water. When we imaged the SWNT–CYT conjugate, we observed many linear objects (SWNTs) with spherical objects along the linear structure (Fig. 5a). The height profile of the hybrid material shows that objects with an average size of 2 nm are spaced along the length of the SWNT which has a height of 1 nm (Fig. 5b and c). The AFM image clearly indicates successful coupling between SWNT and CYT·1 in the CuAAC reaction. To confirm that CYT in the hybrid is covalently linked and is not non-covalently absorbed, we prepared a mixed sample of f-SWNT and CYT·1 without the copper catalyst (Fig. 4d). In the AFM images, it is seen that the SWNT in the mixed sample has a very smooth surface both in the forms of isolated or bundled tubes (Fig. 5e and f). This suggests that the adsorbed proteins were washed off from the sidewall of SWNTs. Therefore, the wire-like structure of SWNT observed in the rough height profile in the AFM image clearly supports the formation of the covalent linkage between SWNT and CYT·1.
 | ||
| Fig. 5 AFM images of (a–c) CYT·1@f-SWNT and (d–f) f-SWNT or f-SWNT with holoCYT. Zoomed views (b and e) and height profiles (c and f) are also shown. | ||
Conclusions
In conclusion, cytochrome b562 with a tethered azide group on the heme propionate side chain was covalently linked to the acetylene moiety introduced on the sidewall of SWNT via a specific covalent bond containing the triazole ring formed by copper-catalyzed click chemistry. This method provides a novel strategy for development of oriented redox-active hemoprotein–SWNT hybrid materials. The present work indicates that our method for fabricating hemoprotein–carbon nanomaterial electrodes has great potential for use in preparing specifically-designed bioelectrode interfaces. Non-covalent conjugation between a genetically incorporated azide-linked amino acid residue of a redox-active copper enzyme and CNT is known to work as an efficient electrical wiring,14 whereas our conjugate via a covalent linkage between SWNT and protein is expected to provide more rigid orientation of the enzymes on the SWNT sidewall. Efforts to expand the attractive features of hemoprotein matrices as electron transfer mediators, sensors, and catalysts using this strategy are now in progress.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (Innovative Areas “Element Block,” Area 2401, KAKENHI 25102527 and 15H00746) from MEXT and the Japan Society for the Promotion of Science (JSPS). A.O. acknowledges support from the Japan Association of Chemical Innovation (JACI). This program was supported by the Strategic International Collaborative Research Program (SICORP), JST. This work was also supported by the JST-ANR program TMOL “Molecular Technology” project MECANO (ANR-14-JTIC-0002-01).Notes and references
- A. Walcarius, S. D. Minteer, J. Wang, Y. Lin and A. Merkoci, J. Mater. Chem. B, 2013, 1, 4878–4908 RSC.
- R. A. S. Luz, A. R. Pereira, J. C. P. de Souza, F. C. P. F. Sales and F. N. Crespilho, ChemElectroChem, 2014, 1, 1751–1777 CrossRef CAS.
- A. de Poulpiquet, A. Ciaccafava and E. Lojou, Electrochim. Acta, 2014, 126, 104–114 CrossRef CAS.
- A. Zebda, C. Gondran, A. Le Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 2, 370 CrossRef PubMed.
- T. Miyake, S. Yoshino, T. Yamada, K. Hata and M. Nishizawa, J. Am. Chem. Soc., 2011, 133, 5129–5134 CrossRef CAS PubMed.
- F. Gao, L. Viry, M. Maugey, P. Poulin and N. Mano, Nat. Commun., 2010, 1, 2 Search PubMed.
- A. T. E. Vilian, V. Veeramani, S. M. Chen, R. Madhu, C. H. Kwak, Y. S. Huh and Y. K. Han, Sci. Rep., 2015, 5, 18390 CrossRef CAS PubMed.
- F. Patolsky, Y. Weizmann and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 2113–2117 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- T. Palacin, H. L. Khanh, B. Jousselme, P. Jegou, A. Filoramo, C. Ehli, D. M. Guldi and S. Campidelli, J. Am. Chem. Soc., 2009, 131, 15394–15402 CrossRef CAS PubMed.
- G. Clavé and S. Campidelli, Chem. Sci., 2011, 2, 1887–1896 RSC.
- I. Hijazi, B. Jousselme, P. Jegou, A. Filoramo and S. Campidelli, J. Mater. Chem., 2012, 22, 20936–20942 RSC.
- D. Guan, Y. Kurra, W. Liu and Z. Chen, Chem. Commun., 2015, 51, 2522–2525 RSC.
- K. Kobayashi, M. Shimizu, T. Nagamune, H. Sasabe, Y. Fang and W. Knoll, Bull. Chem. Soc. Jpn., 2002, 75, 1707–1713 CrossRef CAS.
- D. H. Murgida and P. Hildebrandt, Phys. Chem. Chem. Phys., 2005, 7, 3773–3784 RSC.
- A. E. F. Nassar, W. S. Willis and J. F. Rusling, Anal. Chem., 1995, 67, 2388–2392 CrossRef.
- I. Taniguchi, K. Watanabe, M. Tominaga and F. M. Hawkridge, J. Electroanal. Chem., 1992, 333, 331–338 CrossRef CAS.
- J. Xu, F. Shang, J. H. T. Luong, K. M. Razeeb and J. D. Glennon, Biosens. Bioelectron., 2010, 25, 1313–1318 CrossRef CAS PubMed.
- E. E. Ferapontova, Electroanalysis, 2004, 16, 1101–1112 CrossRef CAS.
- F. W. Scheller, U. Wollenberger, C. Lei, W. Jin, B. Ge, C. Lehmann, F. Lisdat and V. Fridman, Rev. Mol. Biotechnol., 2002, 82, 411–424 CrossRef CAS PubMed.
- N. Bistolas, U. Wollenberger, C. Jung and F. W. Scheller, Biosens. Bioelectron., 2005, 20, 2408–2423 CrossRef CAS PubMed.
- Y. Wu and S. Hu, Microchim. Acta, 2007, 159, 1–17 CrossRef CAS.
- G. X. Ma, T. H. Lu and Y. Y. Xia, Bioelectrochemistry, 2007, 71, 180–185 CrossRef CAS PubMed.
- A. Ramanavicius and A. Ramanaviciene, Fuel Cells, 2009, 9, 25–36 CrossRef CAS.
- L. H. Guo, G. McLendon, H. Razafitrimo and Y. L. Gao, J. Mater. Chem., 1996, 6, 369–374 RSC.
- A. Das and M. H. Hecht, J. Inorg. Biochem., 2007, 101, 1820–1826 CrossRef CAS PubMed.
- H. Zimmermann, A. Lindgren, W. Schuhmann and L. Gorton, Chem.–Eur. J., 2000, 6, 592–599 CrossRef CAS.
- L. Fruk and C. M. Niemeyer, Angew. Chem., Int. Ed., 2005, 44, 2603–2606 CrossRef CAS PubMed.
- C. H. Kuo, L. Fruk and C. M. Niemeyer, Chem.–Asian J., 2009, 4, 1064–1069 CrossRef CAS PubMed.
- M. Sosna, D. Fapyane and E. E. Ferapontova, J. Electroanal. Chem., 2014, 728, 18–25 CrossRef CAS.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- M. Zayats, E. Katz, R. Baron and I. Willner, J. Am. Chem. Soc., 2005, 127, 12400–12406 CrossRef CAS PubMed.
- I. Willner, V. Heleg-Shabtai, R. Blonder, E. Katz, G. Tao, A. F. Bückmann and A. Heller, J. Am. Chem. Soc., 1996, 118, 10321–10322 CrossRef CAS.
- O. A. Raitman, F. Patolsky, E. Katz and I. Willner, Chem. Commun., 2002, 1936–1937 RSC.
- J. C. Vidal, J. Espuelas and J. R. Castillo, Anal. Biochem., 2004, 333, 88–98 CrossRef CAS PubMed.
- M. Pita and E. Katz, J. Am. Chem. Soc., 2007, 130, 36–37 CrossRef PubMed.
- A. Onoda, Y. Kakikura, T. Uematsu, S. Kuwabata and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 2628–2631 CrossRef CAS.
- Y. Kakikura, A. Onoda, E. Kubo, H. Kitagishi, T. Uematsu, S. Kuwabata and T. Hayashi, J. Inorg. Organomet. Polym. Mater., 2013, 23, 172–179 CrossRef CAS.
- A. Onoda, Y. Kakikura and T. Hayashi, Dalton Trans., 2013, 42, 16102–16107 RSC.
- A. Onoda, Y. Ueya, T. Sakamoto, T. Uematsu and T. Hayashi, Chem. Commun., 2010, 46, 9107–9109 RSC.
- K. Oohora, S. Burazerovic, A. Onoda, Y. M. Wilson, T. R. Ward and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 3818–3821 CrossRef CAS PubMed.
- T. Himiyama, A. Onoda and T. Hayashi, Chem. Lett., 2014, 43, 1152–1154 CrossRef CAS.
- K. Oohora, A. Onoda and T. Hayashi, Chem. Commun., 2012, 48, 11714–11726 RSC.
- T. Ono, Y. Hisaoka, A. Onoda, K. Oohora and T. Hayashi, Chem.–Asian J., 2016, 11, 1036–1042 CrossRef CAS PubMed.
- M. Chromiński, K. ó Proinsias, E. Martin and D. Gryko, Eur. J. Org. Chem., 2013, 8, 1530–1537 CrossRef PubMed.
- F. W. J. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra14195a |
| This journal is © The Royal Society of Chemistry 2016 |