
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of fluorination on the surface structure of truxenones†
L. A.
Rochford
*a,
A. J.
Ramadan
 b,
S.
Holliday
c,
T. S.
Jones
a and
C. B.
Nielsen
cd
b,
S.
Holliday
c,
T. S.
Jones
a and
C. B.
Nielsen
cd
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: luke.rochford@warwick.ac.uk
bDepartment of Physics, Clarendon Laboratory, University of Oxford, Oxford OX1 3PU, UK
cDepartment of Chemistry, Imperial College London, South Kensington, SW7 2AZ, UK
dMaterials Research Institute and School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London E1 4NS, UK
First published on 8th July 2016
Abstract
The surface structure of partially fluorinated truxenone (F3-truxenone) molecules on Cu (111) has been probed using a combination of scanning tunneling microscopy (STM) and low energy electron diffraction (LEED). Codeposition of F3-truxenone and the parent truxenone molecule leads to a mix of discrete F3-truxenone and truxenone islands on a Cu (111) surface. Due to the differences in rotational orientation of each type of molecular island proved by LEED the otherwise indistiguishable molecules can be identified in STM images.
Introduction
At its most fundamental, the field of organic electronics relies on interfaces between organic molecules and solid surfaces.1–3 Devices such as organic photovoltaics commonly incorporate at least one interface between an organic molecule and a metal or metal oxide.4,5 As well as control of device performance through interface modification, organic chemistry is widely used to design and modify molecules with device applications in mind.6 Production of electron acceptor materials for the replacement of archetypal acceptors (such as C60) is the subject of huge contemporary research activity.7 Axial substitution of hydrogen for halogen atoms in conjugated small molecules is a widely used method to control their electronic properties.8–10 For example in planar phthalocyanine molecules axial fluorination has been shown to rigidly shift energy levels to lower values while preserving the symmetry of the molecule.11 This shift is of sufficient size to allow fluorinated adducts of the phthalocyanines to accept electrons from electron donating molecules.12 While measurements of the effect of this modification upon relevant molecular energy levels (LUMO/HOMO) are widely undertaken, far less attention has been paid to changes induced in crystal structure – both in single crystals and at interfaces.13,14Forming ordered structures comprised of small conjugated organic molecules on the surface of inorganic crystals is a well-established method of probing their structure and properties.15 Here, we study the effect of partial axial fluorination of truxenone (diindeno[1,2-a; 1′,2′-c]fluorene-5,10,15-trione) (Fig. 1) on surface structure in molecular monolayers. Truxenones have shown particular recent efficacy for use as electron acceptors in organic electronic devices and derivatives have been used to produce highly efficient organic photovoltaic devices.16–18 The use of high resolution scanning tunnelling microscopy (STM) and low energy electron diffraction (LEED) allows the surface structures of truxenone and F3-truxenone to be compared.
 | ||
| Fig. 1 Chemical structures of (a) truxenone and (b) F3-truxenone. | ||
Experimental
Truxenone and F3-truxenone were synthesised following literature procedures19,20 and triply purified by thermal gradient sublimation before degassing 20 °C below the evaporation temperature for several days.21 NMR data and full synthesis details are presented in the ESI (S1 and S2).† A single crystal Cu (111) substrate (Surface Preparation Lab, NL – cutting accuracy 0.1°) was prepared in vacuum by repeated cycles of argon ion sputtering and annealing (Ar+ energy 1.5 keV temperature 550 °C). Standard low-temperature effusion cells (Karl Eberl) were used at 150 °C (truxenone) and 155 °C (F3-truxenone) for molecular growth. Truxenone and F3-truxenone layers were produced by evaporation onto a clean Cu (111) surface, initially monitored using post growth LEED with increasing deposition time. All characterisation was carried out at ambient temperature in a custom-built multi-chamber ultra-high vacuum (UHV) system with a base pressure better than 3 × 10−10 mbar. STM images were recorded with an STM/AFM (Omicron) operated in constant current mode using electrochemically etched polycrystalline tungsten tips. Applied voltages and tunnelling currents are indicated in figure captions. Images and 2D-FFTs were produced using the open source software Gwyddion®. LEED patterns were collected with a SPECTALEED (Omicron) rear-view MCP-LEED with nano-amp primary beam current. Images of these diffraction patterns were captured using a digital CCD camera interfaced to a personal computer, and are presented with their colours inverted for clarity. Simulated patterns and surface meshes were calculated using the open source software LEEDpat4.1.Results and discussion
In order to compare the behaviour of the parent truxenone and the fluorinated derivative (4,9,14-trifluorotruxenone, F3-truxenone) thin films were grown by evaporation in ultra-high vacuum. As truxenone has recently been shown to grow commensurate epitaxial structures on Cu (111) surfaces this surface was selected for growth of both molecules.22 Growth was undertaken in short increments (one minute) until sharp LEED patterns (Fig. 2(d)) were observed (ten minutes in total). The overlayer did not form the same commensurate p(8x8) structure as the parent truxenone, and consisted of twice the number of first order diffraction spots. This is suggestive of rotational domains induced by misalignment of the molecular overlayer mesh with one of the high symmetry directions of the substrate.23 | ||
| Fig. 2 STM images of F3-truxenone/Cu (111) at (a) low (VS = −2 V, IT = 65 pA) and (b) high (VS = −2 V, IT = 65 pA) magnification along with (c) 2D-FFT of the low magnification image. Panel (d) shows a LEED pattern of the surface at 12 eV along with the assigned surface mesh matrix and (e) a simulated pattern (each domain is separately coloured). | ||
STM images were collected from the surface and are presented in Fig. 2(a) and (b). Large domains of F3-truxenone were present on the surface and the molecular visualisation was similar to that observed previously for truxenone. Each F3-truxenone molecule appeared as a three-lobed triangular feature with a less bright central region rather than the single bright triangular shape as previously observed for truxenone.24,25 This additional contouring in the local density of states (LDOS) of the molecule may be due to the fluorine atoms creating an uneven distribution of electron density in the outermost phenyl rings with respect to the core.26,27 Whether this effect is an electronic or local structural effect is problematic to discern with STM measurements. The authors note, however, that we cannot rule out small changes in tip condition being responsible for the change in contrast. The porous honeycomb structure observed was misaligned with the substrate primitive by 26° (±0.5°) according to LEED and STM data (Fig. 2). Some of the ‘pores’ of the honeycomb network appear to be filled and appear with similarly bright contrast to the surrounding molecules in STM images. The identity of these species is unclear but they may be collections of copper ad-atoms (mobile at room temperature), or small impurities around which the layer may have crystallised.
The size of the rhombic molecular unit cell (α = 120°) was the same (r1 = r2 = 2.07 nm) as previously observed in truxenone (within the experimental error) on Cu (111). However, these dimensions coupled with the misalignment with respect to the substrate suggest incommensurate ordering of the F3-truxenone layer. Unlike the unsubstituted truxenone p(8x8) and p(5x5) cases, the translational symmetry of the surface is not preserved as the principle lattice vectors of the overlayer and substrate do not align. With the measured angles and unit cell dimensions the following transfer matrix could be constructed:
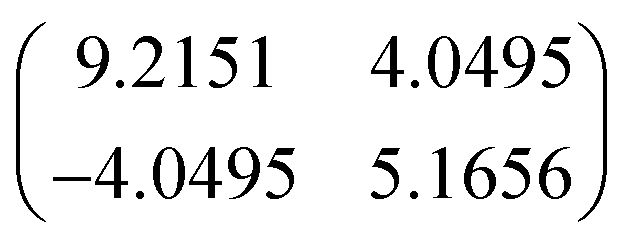
The two dimensional fast Fourier transform (2D-FFT) of the large scale STM image shown in Fig. 2(a) is inset as Fig. 2(c). This shows a single hexagonal pattern, corresponding to one of the mirror domains created by misalignment with a substrate primitive lattice vector. Additional images (see ESI, Fig. S3†) capture regions in which both (symmetry related) orientations are present, and their 2D-FFT reflects the symmetry of the LEED pattern's first order spots. Simulations using the transfer matrix presented are shown in Fig. 2(e) and consider both of these symmetry related domains. The close resemblance of the simulated and experimental LEED pattern confirms that our suggested surface mesh accurately describes the F3-truxenone layer on Cu (111).
Although only a proportion (1/4) of the axial hydrogen atoms are replaced with fluorine the precursor used in the synthesis ensures that only a single diastereomer is formed.28 The surface immobilisation will, as in the case of unsubstituted truxenone, form two enantiomers of the F3-truxenone. Our STM images do not allow discrimination between the enantiomers as single atoms cannot be resolved and no ‘handedness’ is obvious in the molecular footprint.29 Commensurate truxenone structures on Cu (111) can accommodate both enantiomers, and this may also be true with incommensurate structures of F3-truxenone seen here. Although LEED patterns show two domains, the underlying cause of the misalignment with the substrate is unclear. In one case, enantiopure domains may be forming with one enantiomer responsible for each of the domains.30 However, the packing arrangement of the molecules may not be selective to one or the other surface enantiomer and enantiomorphous domains may be present.31,32 In this case the misalignment of the F3-truxenone with the substrate would be intrinsic to the replacement of the hydrogen atoms with fluorine. Unfortunately this is beyond the capabilities of our combined STM and LEED measurements, even the absolute point group symmetry of the molecular overlayer cannot be unambiguously determined.
The precise adsorption configuration site and the orientation of the molecule with respect to the surface could only be directly measured with synchrotron based techniques. Normal incidence X-ray standing wave (NIXSW), and photoelectron diffraction (PhD) would provide some insight into this but these data are not available at present for this system.33,34 Despite these caveats, the surface mesh extracted from LEED and STM remains valid although without a unique solution.
For direct comparison the parent truxenone molecule was evaporated to a similar coverage on Cu (111) and characterisation of this interface is shown in Fig. 3. As previously reported, a porous network was formed with a rhombic unit cell aligned to define a p(8x8) surface mesh.
 | ||
| Fig. 3 STM images of truxenone/Cu (111) at (a) low (VS = −1.25 V, IT = 100 pA) and (b) high (VS = −1.25 V, IT = 100 pA) magnification along with (c) 2D-FFT of the low magnification image. Panel (d) shows a LEED pattern of the surface at 12 eV along with the assigned surface mesh matrix and (e) a simulated pattern. | ||
LEED patterns reflect this ordering and symmetry, and no mirror-plane related domains are observed due to the alignment of the organic overlayer and substrate primitive lattice vectors.
Inspection of STM images of single-component (truxenone or F3-truxenone) films highlights the striking similarity between the appearances of individual molecules. Attempts at bias-dependent imaging to discern differences in molecular orbital derived states were unsuccessful due to the instability of the tip and/or surface at room temperature. In order to compare the observations in STM and crystallography of both molecules simultaneously they were co-evaporated onto Cu (111) from separate evaporation sources. As the flux was being effectively doubled compared to growth of single-component films, the growth time was halved.
Mixed truxenone and F3-truxenone films exhibited LEED patterns which could be understood as a superposition of the individual patterns of each component (Fig. 4). This was strongly suggestive of the formation of discrete domains of each molecule without ‘mixtures’ of the two molecules forming, although small amounts of local co-crystallisation cannot be completely ruled out. We can be sure that no ordered regions greater in size than the LEED transfer width (approx. 50 nm) with structures different to those previously discussed are present. It could be reasonably expected that intermixing molecules which differed only by their axial fluorination would lead to a modification to the unit cell or orientation observed.35
 | ||
| Fig. 4 STM images of co-deposited F3-truxenone and truxenone on Cu (111) at (a) low (VS = −1.30 V, IT = 85 pA) and (b) high (VS = −1.5 V, IT = 75 pA) magnification. A false cover overlay is added to the low magnification image in panel (c) to mark domains of F3-truxenone (green) and truxenone (red). The 2D-FFT of the low-resolution image is shown in (d) along with (e) the LEED pattern at 12 eV and (f) the combined simulated pattern (yellow = truxenone, blue + red = F3-truxenone). | ||
Co-deposited films were also analysed by STM and exhibit familiar open honeycomb structures as observed individually for truxenone and F3-truxenone. On close inspection the rotational orientation of individual islands (assuming that poorly-defined regions are bare Cu substrate) is not the same across the imaged region. This can be clearly demonstrated by taking the 2D-FFT of the STM image, which produced a remarkably close approximation of the LEED pattern from this surface.
STM images can therefore be inspected and otherwise indistinguishable molecules can be identified by their in-plane rotational orientation. To show the local orientation (and therefore the composition of each island), the large-scale STM image in Fig. 4 has been overlaid with false coloured tiles to indicated areas in which F3-truxenone and truxenone have separately crystallised. Lateral heterojunctions of two dimensional inorganic materials (such as graphene and hexagonal boron nitride) have attracted significant recent interest for electronic device applications.36 The results presented here demonstrate that with some fine tuning, organic lateral heterojunctions could be realised.
Conclusions
The surface structure of truxenone and partially fluorinated truxenone on a Cu (111) surface are characterised by STM and LEED. When the molecules are co-evaporated they separately crystallise into discrete islands, and despite very similar appearances in STM images when present individually on Cu (111) the orientation of grains with respect to the substrate allows molecular identity to be discerned. The spontaneous organisation of each molecule into discrete islands demonstrates that axial halogen substitution (intended for electronic modification) can produce concomitant surface structure changes. Further tuning of the molecular structure through synthetic chemistry may allow the degree of separation to be controlled and intermixed phases to be observed. If further control can be demonstrated, the fabrication of lateral molecular heterojunctions could be realised with these molecular systems.Acknowledgements
LAR, AJR and TSJ acknowledge support from the Engineering and Physical Sciences Research Council [Grant Number EP/H021388/1]. SH and CBN acknowledge support from the following funding bodies: EPSRC Project EP/G037515/1, EC FP7 Project SC2 (610115), EC FP7 Project ArtESun (604397), and EC FP7 POLYMED (612538). The raw data associated with this article can be accessed at the following DOI: 10.17632/mmp65j8jgs.2Notes and references
- S. R. Forrest, Chem. Rev., 1997, 97, 1793–1896 CrossRef CAS PubMed.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- N. Koch, ChemPhysChem, 2007, 8, 1438–1455 CrossRef CAS.
- M. T. Greiner, M. G. Helander, W.-M. Tang, Z.-B. Wang, J. Qiu and Z.-H. Lu, Nat. Mater., 2011, 11, 76–81 CrossRef.
- A. Kahn and N. Koch, in The Molecule-Metal Interface, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013, pp. 219–241 Search PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed.
- K. Cnops, G. Zango, J. Genoe, P. Heremans, M. V. Martinez-Diaz, T. Torres and D. Cheyns, J. Am. Chem. Soc., 2015, 137, 8991–8997 CrossRef CAS PubMed.
- P. Sullivan, A. Duraud, L. Hancox, N. Beaumont, G. Mirri, J. H. R. Tucker, R. A. Hatton, M. Shipman and T. S. Jones, Adv. Energy Mater., 2011, 1, 352–355 CrossRef CAS.
- C. B. Nielsen, A. J. P. White and I. McCulloch, J. Org. Chem., 2015, 80, 5045–5048 CrossRef CAS PubMed.
- W. Wu, L. A. Rochford, S. Felton, Z. Wu, J. L. Yang, S. Heutz, G. Aeppli, T. S. Jones, N. M. Harrison and A. J. Fisher, J. Appl. Phys., 2013, 113, 013914 CrossRef.
- J. L. Yang, S. Schumann, R. A. Hatton and T. S. Jones, Org. Electron., 2010, 11, 1399–1402 CrossRef CAS.
- H. Jiang, J. Ye, P. Hu, F. Wei, K. Du, N. Wang, T. Ba, S. Feng and C. Kloc, Sci. Rep., 2014, 4, 7573 CrossRef CAS PubMed.
- J. Endres, I. Pelczer, B. P. Rand and A. Kahn, Chem. Mater., 2016, 28, 794–801 CrossRef CAS.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- C. B. Nielsen, E. Voroshazi, S. Holliday, K. Cnops, B. P. Rand and I. McCulloch, J. Mater. Chem. A, 2013, 1, 73–76 CAS.
- C. B. Nielsen, E. Voroshazi, S. Holliday, K. Cnops, D. Cheyns and I. McCulloch, J. Mater. Chem. A, 2014, 2, 12348–12354 CAS.
- G. Zhang, V. Lami, F. Rominger, Y. Vaynzof and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55(12), 3977–3981 CrossRef CAS PubMed.
- E. V. Dehmlow and T. Kelle, Synth. Commun., 1997, 27, 2021–2031 CrossRef CAS.
- L. Sanguinet, J. C. Williams, Z. Yang, R. J. Twieg, G. Mao, K. D. Singer, G. Wiggers and R. G. Petschek, Chem. Mater., 2006, 18, 4259–4269 CrossRef CAS.
- A. R. McGhie, A. F. Garito and A. J. Heeger, J. Cryst. Growth, 1974, 22, 295–297 CrossRef CAS.
- A. J. Ramadan, C. B. Nielsen, S. Holliday, T. S. Jones, I. McCulloch and L. A. Rochford, RSC Adv., 2016, 6, 17125–17128 RSC.
- L. A. Rochford, I. Hancox and T. S. Jones, Surf. Sci., 2014, 628, 62–65 CrossRef CAS.
- Z.-Y. Yang, Y. Tao, T. Chen, H.-J. Yan and Z.-X. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2105 RSC.
- F. Chen, Z. Hu, Y. Ji, A. Zhao, B. Wang, J. Yang and J. G. Hou, Phys. Chem. Chem. Phys., 2012, 14, 3980 RSC.
- D. G. de Oteyza, A. El-Sayed, J. M. Garcia-Lastra, E. Goiri, T. N. Krauss, A. Turak, E. Barrena, H. Dosch, J. Zegenhagen, A. Rubio, Y. Wakayama and J. E. Ortega, J. Chem. Phys., 2010, 133, 214703 CrossRef CAS PubMed.
- H. Huang, W. Chen and A. T. S. Wee, J. Phys. Chem. C, 2008, 112, 14913–14918 CAS.
- C. Lambert, G. Nöll, E. Schmälzlin, K. Meerholz and C. Bräuchle, Chem.–Eur. J., 1998, 4, 2129–2135 CrossRef CAS.
- W. Xiao, X. Feng, P. Ruffieux, O. Gröning, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2008, 130, 8910–8912 CrossRef CAS PubMed.
- C. B. France and B. A. Parkinson, J. Am. Chem. Soc., 2003, 125, 12712–12713 CrossRef CAS PubMed.
- R. Fasel, M. Parschau and K.-H. Ernst, Nature, 2006, 439, 449–452 CrossRef CAS PubMed.
- F. Stevens, D. J. Dyer and D. M. Walba, Angew. Chem., Int. Ed. Engl., 1996, 35, 900–901 CrossRef CAS.
- D. Woodruff, Surf. Sci. Rep., 2007, 62, 1–38 CrossRef CAS.
- D. P. Woodruff, Prog. Surf. Sci., 1998, 57, 1–60 CrossRef CAS.
- Y. L. Huang, W. Chen and A. T. S. Wee, J. Am. Chem. Soc., 2011, 133, 820–825 CrossRef CAS PubMed.
- M. P. Levendorf, C.-J. Kim, L. Brown, P. Y. Huang, R. W. Havener, D. A. Muller and J. Park, Nature, 2012, 488, 627–632 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional STM images and 2D-FFTs. See DOI: 10.1039/c6ra14158g |
| This journal is © The Royal Society of Chemistry 2016 |