
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel quinoxaline based chemosensors with selective dual mode of action: nucleophilic addition and host–guest type complex formation†
Marium
Ishtiaq‡
a,
Iqra
Munir‡
a,
Mariya
al-Rashida
b,
Maria
c,
Khurshid
Ayub
c,
Jamshed
Iqbal
d,
Ralf
Ludwig
ef,
Khalid Mohammed
Khan
 a,
Syed Abid
Ali
*a and
Abdul
Hameed
*a
a,
Syed Abid
Ali
*a and
Abdul
Hameed
*a
aH. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan. E-mail: abid.ali@iccs.edu; abdul.hameed@iccs.edu; Fax: +92-21-3481901; Tel: +92-21-99261701-2
bDepartment of Chemistry, Forman Christian College (A Chartered University), Ferozepur Road-54600, Lahore, Pakistan
cDepartment of Chemistry, COMSATS Institute of Information Technology, Abbottabad, KPK, Pakistan 22060
dCenter for Advanced Drug Research, COMSATS Institute of Information Technology, Abbottabad 22060, Pakistan
eLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
fDepartment of Physical Chemistry, University of Rostock, Dr.-Lorenz-Weg 1, 18059 Rostock, Germany
First published on 22nd June 2016
Abstract
New quinoxalinium salts 1–5 have been exploited as chemosensors via naked eye, UV-Vis absorption, fluorescence quenching and 1H NMR experiments. New sensors 1–5 showed a dual mode, nucleophilic addition and a host–guest type complex towards anion (F−, AcO− and ascorbate) detection. Small anions (F−/AcO−) showed nucleophilic addition at the C2 position of the quinoxalinium cation, while larger anions (ascorbate), revealed the formation of a host–guest type complex due to the steric hindrance posed by the C3 of the phenyl ring. Nucleophilic addition of small anions (F−/AcO−) leads to the de-aromatization of the quinoxalinium cation. However in the case of the larger anion, ascorbate, the host–guest type complex formation induces changes in the absorption/fluorescence signals of the quinoxalinium moiety. This selective binding has been confirmed on the basis of the 1H NMR spectroscopic technique, whereupon nucleophilic addition of small anions (F−/AcO−) was confirmed by monitoring the characteristic proton NMR signals of Ha and the methylene protons (CH2), which were clearly shifted in the cases of fluoride and acetate ion addition confirming the de-aromatization and nucleophilic addition. Whereas no such peak shifting was observed in the case of ascorbate ion addition confirming the non-covalent addition of ascorbate. Theoretical insight into the selectivity and complexation behavior of the ascorbate ion with the quinoxaline moiety is gained through density functional theory (DFT) calculations. Moreover, the absorption properties of these complexes are modeled theoretically, and compared with the experimental data. In addition, the thermal decomposition of sensors (1 and 2) has been studied by the means of differential scanning calorimetry (DSC), thermogravimetry (TG), and differential thermogravimetry (DTG) to signify their utility at variable temperatures.
Introduction
Anion sensing via chemosensors as receptors carries great importance in observing biological processes, clinical diagnostics, and environmental monitoring. A number of compounds as sensors have been reported for different anions such as halides, acetate, phosphate, and citrate etc. in regulating their certain levels in biological processes and variable ecosystems.1 Among the halides, fluoride (F−) continues to receive much attention due to its wide spread applications in dental care, and its role in certain enzyme inhibitions.2–4 Similarly, acetate ions (AcO−) play an important role in many metabolic processes. It has also been known to serve as a tracer for certain malignancies. Acetate has been widely studied in prostate cancer and metastases.5–7 Ascorbic acid (vitamin C) is an essential nutrient in humans and animals; it serves as an important co-factor in various enzymatic reactions.8,9 Ascorbic acid is one of the important biological molecules that are responsible for maintaining cell redox homeostasis. Imbalances in redox status lead to oxidative stress and many pathophysiological disorders including aging and cancer.10 Tumors have been known to maintain a reduced microenvironment, consequently oxidized and reduced forms of hyperpolarized 13C-labeled ascorbic acid (vitamin C) have been used as probes to examine tumor redox status.11 Therefore, vitamin C determination as an analyte presents an interesting biological aspect.Ion detection, including small and large analytes, is an exciting domain to regulate their concentration at a certain level in different biological systems, food, drinking water processes, and industrial and clinical analysis etc.12–14 Traditional methods of ion detection, such as spectroscopy, chromatography and ion selective electrodes suffer from limitations when it comes to biomedical research applications particularly in the imaging of cells and tissues.15 The use of fluorescent ion probes offers alleviation from these drawbacks and aims to provide a more efficient means of cell imaging, combining selectivity with ease of detection.16–20 Some selected receptors (probes) for different ion detection have been presented in Table S1.† Although quite a few fluorescent ion probes have been known,1 the interest in the quest to design new, even more efficient probes with possible biological imaging applications continues to increase worldwide. In the present study, we synthesized quinoxaline based compounds and employed them as chemosensors for different analytes, including F−, AcO−, and ascorbate. The anion sensing results revealed the dual mode (nucleophilic addition and host–guest interaction) of quinoxaline based probes for small and large analytes. The quinoxaline scaffold is an important biological template to assemble different molecules of diverse applications especially in the pharmaceutical industry.21 The known medicinal significance and important role as a pharmacophore in many drugs makes the quinoxaline moiety a fascinating template for the synthesis of ion sensing molecules (Fig. 1).
 | ||
| Fig. 1 Quinoxaline based probes 1–5 and F−, AcO−, and ascorbate sensing (naked eye view). | ||
Results and discussion
The probes (receptors) are generally known to interact with analytes (anions) either via covalent binding, involving addition22 or substitution type reactions,23,24 or via non-covalent interactions that involve the formation of host–guest type complexes.25–27 A recent study with a quinoline based probe has described the deprotonation mechanism with fluoride and acetate anions to induce changes in the observed absorption/fluorescence pattern.24 Another anion sensing study with 9-alkoxy acridinium has demonstrated the nucleophilic addition of anions (F−/AcO−) at the C9 position of the acridinium ring causing de-aromatization, consequently inducing drastic changes in the absorption/fluorescence signals of the acridinium moiety.22 Our studies with quinoxalinium salts 1–5 revealed a dual mode of action, that is; (1) via nucleophilic addition of small anions (F−/AcO−), and (2) via host–guest type complex formation with larger anions (ascorbate). This different mode of binding with small and large anions serves to establish selectivity among anion sensing. Moreover, the basicity of the anions (F− > AcO− > ascorbate) further adds to the selective pattern of receptors 1–5.Nucleophilic addition of anions to the quinoxalinium salts 1–5 at the electron deficient position C2 seems feasible. For smaller anions (F−, AcO−), there is less steric hindrance posed by the C3 phenyl ring. In order to further explore the reactivity and selectivity of the quinoxalinium salts towards F−, AcO−, and ascorbate, five derivatives have been synthesized according to Scheme 1. Synthesis was carried out starting from commercially available acetophenones in three steps; (i) oxidation, (ii) coupling with o-phenylenediamine, and (iii) N-alkylation to afford the desired quinoxalinium salts 1–5, oxidation of the α-methyl group of acetophenone was accomplished using selenium oxide. The resultant dicarbonyl 7 was then coupled with diamine 8 to furnish the corresponding quinoxaline derivatives.28N-Alkylation of the quinoxaline analogues was carried out by using ethyl iodide at 120–124 °C in toluene to produce the desired quinoxalinium salts 1–5 (Scheme 1). The alkylation reaction was found to be regioselective, even in an excess (>10 equiv.) of ethyl iodide only N1 was alkylated. This offers additional benefits in anion sensing as the nucleophile (the analyte anion) will only attack the C2 position of the quinoxalinium salt.
 | ||
| Scheme 1 Synthetic layout of quinoxaline-based probes 1–5. | ||
The addition of the anions (F−, AcO− and ascorbate) was also studied using 1H NMR spectroscopy. The nucleophilic addition of F−/AcO− to probe 1 and 2 was confirmed by the shifting of the characteristic 1H NMR signals, while in the case of the ascorbate anion, no change in the NMR peaks was observed even after the addition of an excess (>30 equivalent) of ascorbate. This is due to the large size of the ascorbate molecule that interacts with the probes via a host–guest mode to induce changes in the absorption/fluorescence bands (Fig. 5). The characteristic 1H NMR signals (Ha and CH2) of probe 2 were monitored after adding different equivalents of F− anions (1–5 equiv.). Upon addition of 1 equivalent of fluoride ions, the NMR spectra showed the disappearance of a singlet peak for the Ha proton at δ 9.71 with the concomitant appearance of a new peak at δ 5.99 that confirms the nucleophilic addition of anion. Moreover, the CH2 proton signal (quartet) (originally at δ 5.19) was also shifted up-field as a multiplet at δ 3.72 (Fig. 2). Similar patterns of NMR peak (Ha and CH2) shifting upon addition of F− ions were observed for probe 1, the only difference being the complete disappearance of a singlet signal for the Ha proton (originally at δ 9.70), after the addition of 5 equivalents of F− anions. Subsequently, the multiplet for the methylene group (CH2) shifted up-field at δ 3.68 (Fig. S2†). For the acetate ion, the pattern of its nucleophilic addition to the C2 position of probe 2 was found to be identical to that of F− ion addition, however, the complete disappearance of the Ha peak (originally at δ 10.27) was observed after the addition of 5 equivalents of ammonium acetate, which can be attributed to the difference in the size of the anion (AcO− > F−) (Fig. 2). The photospectroscopic and 1H NMR results clearly demonstrate that small anions F− and AcO− follow the nucleophilic addition mode, whereas for the larger anion ascorbate, observed absorption/fluorescence changes occurred due to collision quenching in the host–guest type complex.25,29

 | ||
| Fig. 2 Study of anion (F−/AcO−) addition to probe 2via1H NMR spectra; (A) (a) without F− anion and (b) with 1 equiv. of F− source (TBAF) in CD3CN as the solvent; (B) (a) without AcO− anion and (b) with 5 equiv. of ammonium acetate in DMSO-d6 as the solvent. | ||
The spectroscopic properties of the quinoxalinium probes 1–5 for anion (F−, AcO−, ascorbate) sensing via “switch off” response were studied using the naked eye, UV-visible, fluorescence and 1H NMR analysis. A typical probe 2 having an electronegative 4′-fluoro substituent at the C3 phenyl ring, showed characteristic absorption bands at 262, 290, and 368 nm and blue-green fluorescence at 570 nm with a shoulder band at 480 nm in acetonitrile as the solvent (Fig. 3). While probe 1 bearing a 4′-methyl substituent showed two slightly shifted bands at 242, 296 nm and a red shift at 374 nm (Fig. 4). In the case of probe 1 a yellow-red fluorescence signal appeared at 670 nm. Details of the absorption/fluorescence signals for all probes 1–5 have been summarized in Table 1.
 | ||
| Fig. 3 (a) (i) Naked eye view (ii) under UV light at 254 nm; (b) absorption spectra of probe 2; (c) fluorescence spectra (excitation 278 nm); (d) fluorescence spectra (excitation 380 nm) (5 × 10−5 mM) – upon systematic addition (0.5–5 equiv.) of TBAF as a fluoride (F−) source in acetonitrile as the solvent. | ||
 | ||
| Fig. 4 (a) (i) Naked eye view (ii) under UV light at 254 nm; (b) absorption spectra of probe 1; (c) fluorescence spectra (excitation 278 nm); (d) fluorescence spectra (excitation 380 nm) (5 × 10−5 mM) – upon systematic addition (0.5–5 equiv.) of TBAF as a fluoride (F−) source in acetonitrile as the solvent. | ||
| Probes | Absorptions bands (nm) | Fluorescence (ex: 278 nm) | ||
|---|---|---|---|---|
| 1 | 242 | 296 | 374 | 570, 670 |
| 2 | 262 | 290 | 368 | 475, 570 |
| 3 | 269 | 290 | 354 | 550 |
| 4 | 245 | 292 | 360 | 550 |
| 5 | 260 | 290 | 365 | 550 |
The fluorescence sensing profile of probes 1–5 to determine biologically important anions (F−, AcO−, and ascorbate) was studied in acetonitrile/DMSO to overcome the solubility issues. Tetrabutylammonium fluoride (TBAF) was used as a source of F−, while ammonium acetate and sodium L-ascorbate salts were used as sources for AcO− and ascorbate anions respectively. The solutions, made in DMSO for AcO− and ascorbate determination, were further diluted with acetonitrile for absorbance/fluorescence (5 × 10−5 mM) studies. For all the investigated anions, a decrease in fluorescence signal “switch off” was observed upon anion addition. Results showed that with small anions (F−/AcO−) fluorescence quenching occurred due to the de-aromatization of the quinoxalinium ring (Fig. 2), while the large analyte (ascorbate) quenches fluorescence via a photo-induced electron transfer (PET) mechanism (Fig. 5).29 PET is an excited state electron transfer process from the donor to the excited state of the fluorophore moiety.12
 | ||
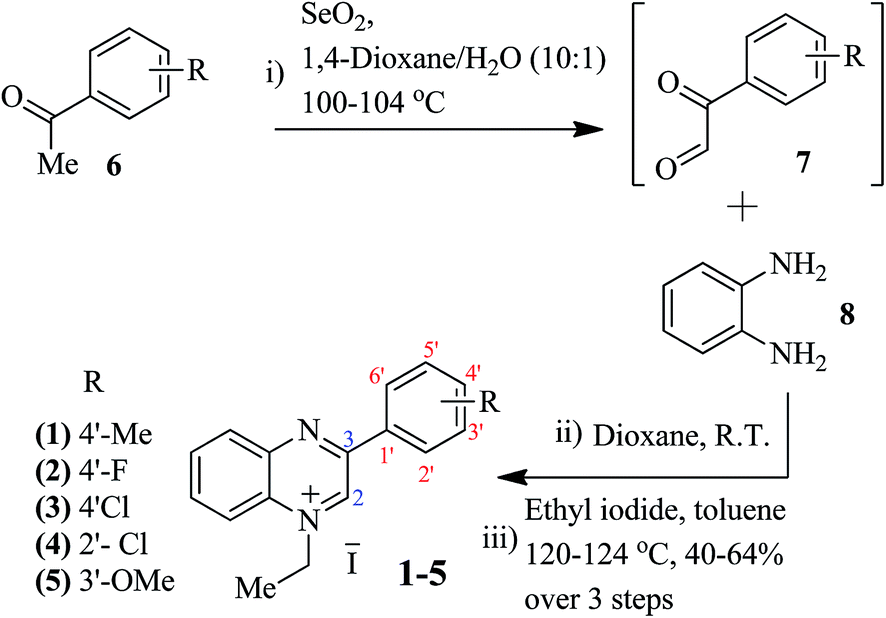
| Fig. 5 Ascorbate addition study with probe 2: (a) absorption spectra in CH3CN indicating a decrease in absorption intensity at 378 nm with a systematic increase in the addition of ascorbate ions (1–30 eq.), and (b) the reduced absorption intensity at 378 nm is replenished upon subsequent additions of methanol, suggesting a reversible host–guest type interaction. Note: stock solutions prepared in DMSO and then diluted in CH3CN or MeOH (5 × 10−5 mM). | ||
A gradual quenching of fluorescence emission intensity at 570 and 670 nm was observed for representative probes 2 and 1 respectively after incremental additions (0.5, 1, 5⋯10 equivalent) of F− anions (TBAF). With the addition of 5 equivalents of TBAF, the fluorescence intensity (excitation at 278) of both probes 2 (Fig. 3) and 1 (Fig. 4) was quenched to a large extent with a concomitant increase at 475 nm (Fig. 3c). Upon excitation at 380 nm the fluorescence quenching follows the same pattern for F− addition (0.5, 1, 5⋯10 equivalent), however, a shift of fluorescence signal was observed, from 570 to 770 nm (Fig. 3d). Fluorescence quenching of probe 1 with the ascorbate anion (Fig. S3c and d in ESI†) follows the same pattern as for F−; it was, however, found to be less efficient in the case of the AcO− anion even after the addition of 10 equivalents of ammonium acetate (Fig. S3a and b in ESI†). Interestingly, in the case of the highly electronegative fluoride substituent containing probe 2, acetate sensing results were found to be better where fluorescence intensity almost diminished in the presence of 10 equivalents of ammonium acetate (Fig. S1b†). The UV-Vis absorption peaks for both of the quinoxalinium salts (1 and 2) were drastically decreased upon the addition of F− anion from 0–5 equivalents, as observed by the color change from light brown to light yellow which remained constant after the addition of 5 equivalents of F− anions (Fig. 3 and 4). These characteristic changes in color, intensity of absorption bands, and fluorescence emission strongly suggest the de-aromatization of the quinoxaline ring as a result of small anion (F−/AcO−) addition, or collision quenching22 in the case of the large analyte (ascorbate) via host–guest type complex formation (Fig. 5). Ascorbate interaction via the formation of a host–guest type complex with the quinoxalinium cation was further confirmed by adding methanol to the same fluorescence quenched solutions of the probes (in DMSO) containing ascorbate ions, whereby previously diminished absorbance was replenished, as indicated by an increase in the absorbance at 378 nm (Fig. 5b).
The absorption/fluorescence study was further extended with three more quinoxalinium salts 3–5 having 4′- or 2′-chloro, and 3′-methoxy substituents at the phenyl ring, to observe their effect in anion sensing process. Fluoride ion determination patterns with 4′-chloro (probe 3) and 2′-chloro (probe 4) substituted quinoxalinium salts were found to be similar to the ones described for probes 1 and 2. Upon 5 equivalent additions of F−, all characteristic absorption peaks (Table 1) disappeared (Fig. S5†). In the fluorescence quenching experiments, probe 3 showed a rapid decline, while probe 4 with the 2′-Cl substituent showed a more or less 50% fluorescence quenching which could be due to the steric effects of the 2′-chloro substituent. Moreover, with the AcO− and ascorbate anions absorption/fluorescence quenching was less diminished even after adding 20 equivalents of the corresponding anion which strengthens the steric congestion factor for the 2′-chloro substituent (Fig. S6†). Probe 5 bearing a 3′-OMe group at the phenyl ring showed a decline in absorption peak with 5 equivalent additions of TBAF, while fluorescence quenching >90% was obtained after 10 equivalent additions of the F− source. This could be due to the steric factor of the 3′-methoxy group at the phenyl ring in comparison to the 4′-position probes. Consequently, the results of the absorption/fluorescence quenching with AcO− and ascorbate anions were less prominent which is in accordance with the fact that steric congestion around the reactive position has a significant effect on the anion sensing process (Fig. S7†). Moreover, thermal studies of representative probes (1 and 2) have been performed to elaborate the stability and decomposition temperatures of the quinoxalinium salts. Thermal analysis provides additional information about molecular interactions and inclusion behaviour of the chemosensors with guest molecules.30–32
Computational studies
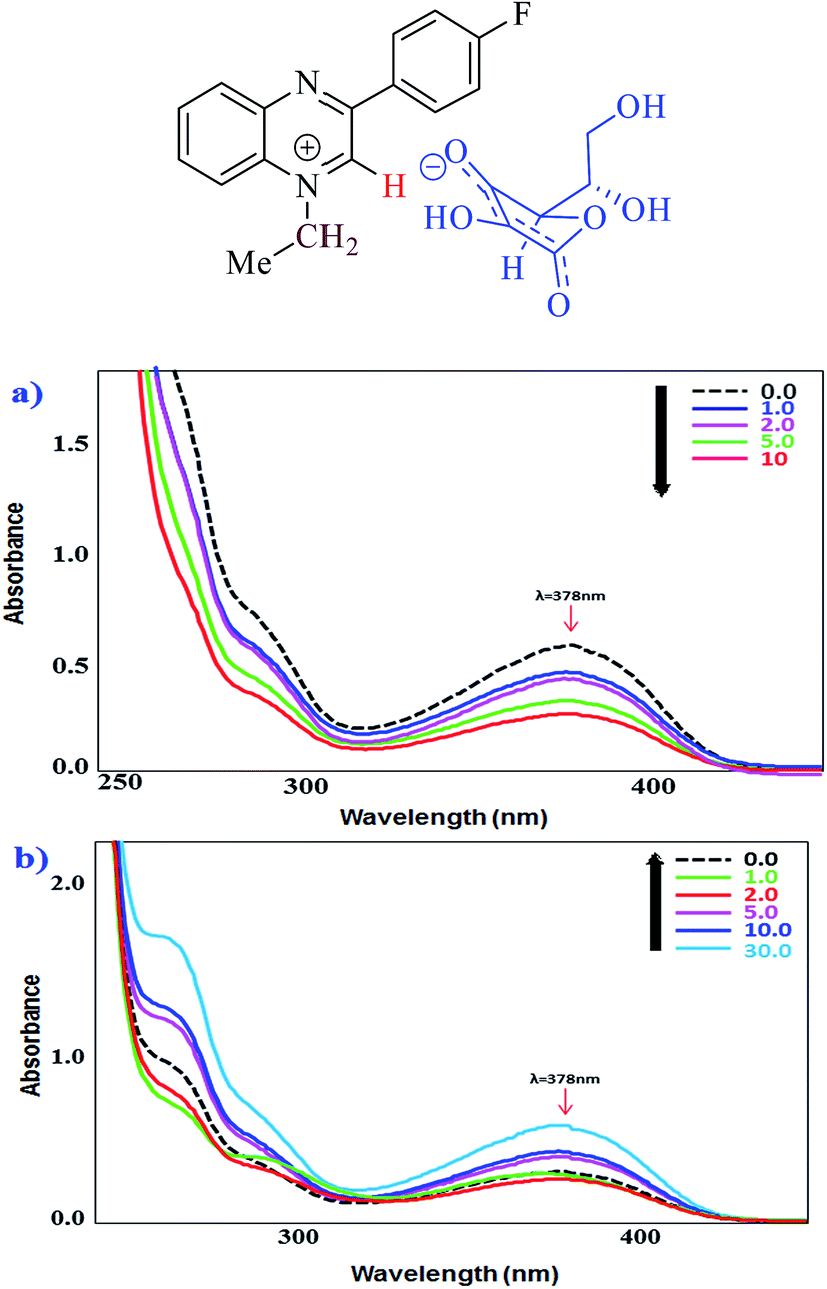
Density functional theory (DFT) calculations have been performed to gain insight into the complexation of the ascorbate anions with the quinoxaline based probes, and the effect on the UV-Vis absorption spectra. The experimental data suggest that the ascorbate ion replaces the iodide through a collision therefore, the ascorbate ion is held with the quinoxaline skeleton through non-bonding interactions. For non-bonding interactions, the ascorbate ion can orient on the quinoxaline skeleton in a variety of ways. Moreover the complexity increases further with a number of conformations being possible for the ascorbate ion. Fortunately, the information regarding the stable conformations of ascorbic acid and the ascorbate ion are already described in the literature.33,34 According to these reports, global minimum geometry is shown in (Fig. 6a). We have not only considered this global minimum geometry in our study but, we have also considered another local minimum (shown in Fig. 6b). This local minimum differs from the global minimum only in one intramolecular non-bonding interaction. The O6–H acts as a hydrogen bond donor to O5 in the global minimum whereas the same O6–H is a hydrogen bond donor to O3 in the local minimum.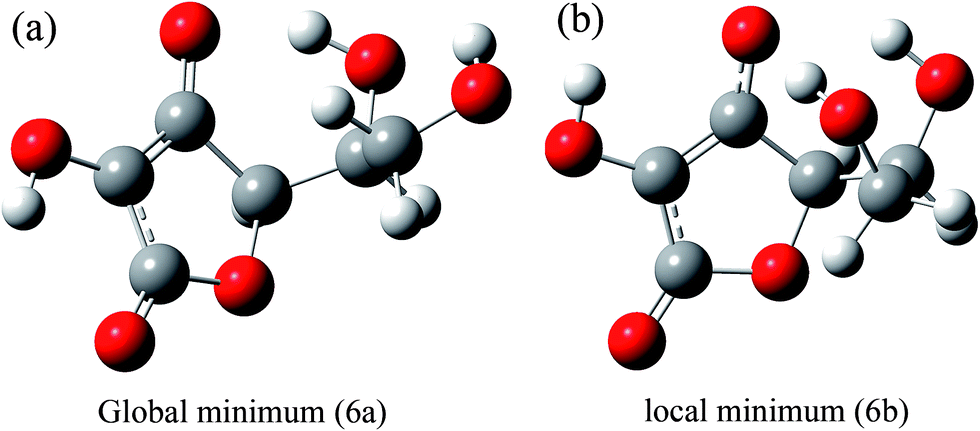 | ||
| Fig. 6 The global 6a and local minimum 6b geometries of the ascorbate ion. | ||
Considering these geometries for the ascorbate ion, we have obtained a number of geometries for the non-bonding interactions of the ascorbate ion with the quinoxaline based cation. The optimized geometries of these complexes (7a–7e) are shown in Fig. 7. The binding energies between the two fragments and relative energies with respect to the most stable isomer are given in Table 2. The complex 7b (the isomer with the lowest energy) is not the complex with the highest binding energy. The highest binding energy is calculated for 7d.
 | ||
| Fig. 7 Optimized geometries of the ascorbate quinoxaline complexes 7a–7e. | ||
| Entry | E rel | E bind (kcal mol−1) | UV-Vis 1 (epsilon) | UV-Vis 2 (epsilon) |
|---|---|---|---|---|
| 7a | 4.82 | −82.64 | 367 (9100) | 763 (750) |
| 7b | 0.00 | −87.46 | 353 (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700) 700) |
763 (260) |
| 7c | 8.85 | −81.92 | 350 (10700) |
761 (2400) |
| 7d | 1.01 | −89.76 | 388 (9200) | 756 (50) |
| 7e | 3.61 | −87.17 | 368 (7000) | 724 (50) |
All of these optimized complexes are characterized by two absorption bands: a quite intense absorption band between 250 and 400 nm, and a very weak absorption band between 700 and 800 nm. The latter absorption band (although weak) is a characteristic band for non-covalent interactions between ascorbate and quinoxaline moieties. This absorption band was missing in an optimized geometry in which the ascorbate moiety binds covalently to the quinoxaline skeleton. The existence of absorption bands between 650 and 800 nm in the experimental spectrum (see ESI-S1†) confirms the existence of a non-covalent complex and also proves the phenomenon of collision quenching.
After establishing through experiment and theory that the interaction between the ascorbate and quinoxaline is non-bonded in nature, attention is paid to the intensity of the absorption band between 250 and 400 nm. From Fig. 8, it is quite obvious that the intensities of absorption bands for 2a and 2b (iodo complexes) are relatively higher than the intensities for 7a to 7e (ascorbate complexes). Comparison of the experimental and theoretical data also suggests that a number of geometries exist for 1:1 interactions between the ascorbate and quinoxaline moieties. This inference is based on the appearance of a number of peaks between 700 and 800 nm (Fig. S1c†). Each complex is believed to give rise to one peak in the region between 700 and 800 nm, as evident from the theoretical calculations.
 | ||
| Fig. 8 Comparative theoretical absorption spectra for the ascorbate–quinoxaline complexes (7a–7e) and iodo quinoxaline (2a–2b), calculated at CAM-B3LYP/6-31G(d,p). | ||
The effect of increasing the concentration of the ascorbate ions does not affect the UV-Vis absorption spectrum between 250 and 400 nm; however, the changes in the region between 600 and 800 nm are indicative of changes in the geometries of the complexes. It might be possible that a number of ascorbate ions per unit of quinoxaline increases which significantly affects the absorptions in this region.
Conclusion
In conclusion, quinoxalinium salts 1–5 have been explored as chemosensors for the detection of three biologically important anions (F−, AcO−, and ascorbate) via naked eye, UV-visible absorption, fluorescence quenching, and 1H NMR spectroscopic techniques. In the case of the small anions (F−/AcO−), nucleophilic addition at the C2 position leads to de-aromatization of the quinoxaline ring, while the large ascorbate molecule formed a host–guest type complex. This selective binding has been confirmed on the basis of the 1H NMR spectroscopic technique, whereupon covalent/nucleophilic addition of small anions (F−/AcO−) was confirmed by monitoring the characteristic proton NMR signals of Ha and methylene (CH2), which were clearly shifted in the case of fluoride ion and acetate ion addition confirming the de-aromatization and nucleophilic addition. Whereas no such peak shifting was observed in the case of ascorbate ion addition confirming the non-covalent addition of the ascorbate. Moreover, the thermal stability studies of probes 1 and 2 have been carried out to broaden the applicability range of these quinoxaline-based probes (1–5) at variable temperatures. Results from the theoretical (DFT) calculations are supportive of a collision quenching mechanism. Computationally calculated UV/vis absorption intensities, for non-bonded ascorbate–quinoxaline complexes are lower than the corresponding absorption intensities for covalently bonded iodo-quinoxaline, which is consistent with the experimental finding. Comparison of the theoretical and experimental UV-Vis spectra also suggests that a number of ascorbate quinoxaline complexes exist under the experimental conditions. Moreover, the nature of complexation is believed to change upon changing the concentration of ascorbate ions.Materials and methods (chemistry)
Different starting materials which include substituted acetophenone (≥99%), selenium dioxide (99.9%), 1,2-diaminobenzene (≥99%), tetrabutylammonium fluoride trihydrated (TBAF) (98%), ethyl iodide (99%), ammonium acetate (≥98%), and sodium L-ascorbate (≥98%) were purchased from Sigma-Aldrich and used without any purification unless otherwise stated. The analytical grade solvents such as 1,4-dioxane, ethyl acetate, diethyl ether and hexane were used as solvents for preforming reactions, and also for purification purposes. Distilled water was used as the solvent for reaction workup purposes. All the reactions were performed in oven dried round bottom flasks. Thin layer chromatography (TLC) was used to monitor the reaction using silica gel 60 aluminium-backed plates 0.063–0.200 mm as the stationary phase with analytical grade solvents such as ethyl acetate (EtOAc), diethyl ether, n-hexane etc. Chromatograms were visualized with UV light (254 nm) or using different staining mixtures such as basic potassium permanganate, vanillin, and bromocresol green. Infrared spectra IR (KBr discs) were recorded between 4000 and 500 cm−1 on a Bruker Vector-22 spectrometer. TGA and DSC analyses were carried out on a TA instrument, model number SDT Q600. Aluminum heating/sample pans were used. The sample weight used was 3–6 mg. Decomposition profiles were obtained at different constant heating rates, that is, 20 °C min−1. All analyses were carried out under a nitrogen atmosphere (flow rate was 100 mL min−1). The data was processed by use of Universal Analysis 2000 software, version 4.2E (TA Instruments, USA), and MS Excel 2013. Bruker spectrometers were used to record 1H NMR spectra at 300, 400 or 500 MHz and 13C NMR at 75, 100 or 125 MHz as dilute solutions in the appropriate deuterated solvent at 25 °C.All the chemical shifts were recorded on the δ-scale (ppm) using residual solvents as an internal standard (DMSO; 1H 2.50, 13C 39.43 and CHCl3; 1H 7.26, 13C 77.16). Coupling constants were calculated in Hertz (Hz) and multiplicities were labelled as s (singlet), d (doublet), t (triplet), q (quartet), or quint (quintet) and the prefixes br (broad) or app (apparent) were used. Mass spectra (ESI+) were recorded at Finnigan MAT-321A, Germany by using electrospray (ES+), electron impact (EI+) or FAB (fast atom bombardment) techniques. Melting points of solids were determined using a Stuart™ melting point apparatus SMP3 apparatus and are uncorrected. All the absorbance measurements were made on an Ultrospec 4300 pro UV/visible spectrophotometer (GE Healthcare, Germany) using quartz cells of 10 mm path length. The fluorescence spectra were scanned at 25 °C on a Spectromax 5 Spectrofluorimeter (Molecular Devices, USA), using 96 well standard fluorescence plates (Nunc™, Germany). The emission range was selected as 300 to 800 nm, with an excitation at wavelength 278 and 380 nm. For anion sensing, stock solutions of probes 1–5 were made in CH3CN or DMSO and then diluted CH3CN (5 × 10−5 mM). Different anion sources (TBAF, NH4OAc, and sodium ascorbate) were added sequentially up to 30 equivalences.
General procedure for the synthesis of the quinoxalinium salts (1–5)
:1, v/v). To this solution, selenium dioxide was added (5.59 mmol, 1.5 equiv.) at room temperature and then heated at 100 °C for 3 h until no starting material was observed using TLC analysis. The reaction mixture was cooled to room temperature and then the excess of selenium was removed using vacuum filtration. The resulting filtrate was concentrated in vacuo and subsequently treated with an excess of water (25 mL) at high temperature (100 °C). It was then treated with activated charcoal to afford the corresponding dicarbonyl intermediate 7 as a white solid and was used without purification in the next step.
Computational methodology
All calculations were performed using the Gaussian 09 suite of programs.35 DFT methods were used to investigate the geometries and electronic properties of the molecules.36,37 The geometries of all the molecules were optimized with the Coulomb attenuated method (CAM), a hybrid functional that merges the qualities of long range correction, of B3LYP for calculation of excitation energies with the 6-31G(d,p)38 basis set. The excited state geometries were calculated with TD-DFT at TD-CAM-B3LYP/6-31G(d,p)39–41 level of theories, in the gas phase. The solvent effect was studied through the polarization continuum model (PCM).Acknowledgements
We are thankful to the Higher Education Commission (HEC) of Pakistan (Project No. 20-2073) for providing financial support for this project. IM was the recipient of the indigenous Ph. D. scholarship from the Higher Education Commission (HEC) of Pakistan.References
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- M. Kleerekoper, Endocrinol. Metab. Clin. North Am., 1998, 27, 441–452 CAS.
- K. L. Kirk, Biochemistry of inorganic fluoride, Springer, 1991 Search PubMed.
- R. Săndulescu, E. Florean, L. Roman, S. Mirel, R. Oprean and P. Suciu, J. Pharm. Biomed. Anal., 1996, 14, 951–958 CrossRef.
- F. P. Kuhajda, E. S. Pizer, J. N. Li, N. S. Mani, G. L. Frehywot and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3450–3454 CrossRef CAS.
- D. Sharma, S. A. Kumar and S. K. Sahoo, Tetrahedron Lett., 2014, 55, 927–930 CrossRef CAS.
- J. Czernin, M. R. Benz and M. S. Allen-Auerbach, PET Clinics, 2009, 4, 163–172 CrossRef PubMed.
- C. Tsao, in Vitamin C in health and disease, ed. L. Packer and J. Fuchs, Marcel Dekker Inc, New York, 1997, pp. 25–58 Search PubMed.
- S. Bsoul and G. Terezhalmy, J. Contemp. Dent. Pract., 2004, 5, 1–13 Search PubMed.
- M. Paidi, J. Schjoldager, J. Lykkesfeldt and P. Tveden-Nyborg, Nutrients, 2014, 6, 1809 CrossRef CAS PubMed.
- S. E. Bohndiek, M. I. Kettunen, D.-e. Hu, B. W. Kennedy, J. Boren, F. A. Gallagher and K. M. Brindle, J. Am. Chem. Soc., 2011, 133, 11795–11801 CrossRef CAS PubMed.
- A. P. De Silva, H. N. Gunaratne, T. Gunnlaugsson, A. J. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R53 CrossRef CAS.
- S.-X. Wang, Z.-H. Wang, X.-T. Cheng, J. Li, Z.-P. Sang, X.-D. Zhang, L.-L. Han, X.-Y. Qiao, Z.-M. Wu and Z.-Q. Wang, Environ. Health Perspect., 2007, 643–647 CrossRef CAS PubMed.
- X. Zhu, J. Wang, J. Zhang, Z. Chen, H. Zhang and X. Zhang, Sensors, 2015, 15, 1611–1622 CrossRef CAS PubMed.
- Q. Jin, L. Feng, D.-D. Wang, Z.-R. Dai, P. Wang, L.-W. Zou, Z.-H. Liu, J.-Y. Wang, Y. Yu and G.-B. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 28474–28481 Search PubMed.
- G. Sivaraman and D. Chellappa, J. Mater. Chem. B, 2013, 1, 5768–5772 RSC.
- T. Anand, G. Sivaraman, M. Iniya, A. Siva and D. Chellappa, Anal. Chim. Acta, 2015, 876, 1–8 CrossRef CAS PubMed.
- C. Arivazhagan, R. Borthakur, R. Jagan and S. Ghosh, Dalton Trans., 2016, 45, 5014–5020 RSC.
- K. M. Vengaian, C. D. Britto, G. Sivaraman, K. Sekar and S. Singaravadivel, RSC Adv., 2015, 5, 94903–94908 RSC.
- J. A. Pereira, A. M. Pessoa, M. N. D. S. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, Eur. J. Med. Chem., 2015, 97, 664–672 CrossRef CAS PubMed.
- Y.-C. Lin and C.-T. Chen, Org. Lett., 2009, 11, 4858–4861 CrossRef CAS PubMed.
- F. Zhao, X. Wu, M. Wang, Y. Liu, L. Gao and S. Dong, Anal. Chem., 2004, 76, 4960–4967 CrossRef CAS PubMed.
- U. N. Yadav, P. Pant, D. Sharma, S. K. Sahoo and G. S. Shankarling, Sens. Actuators, B, 2014, 197, 73–80 CrossRef CAS.
- A. Metzger and E. V. Anslyn, Angew. Chem., Int. Ed., 1998, 37, 649–652 CrossRef CAS.
- X. Shang, Z. Yang, J. Fu, P. Zhao and X. Xu, Sensors, 2015, 15, 28166 CrossRef PubMed.
- T. Gunnlaugsson, A. P. Davis, J. E. O'Brien and M. Glynn, Org. Lett., 2002, 4, 2449–2452 CrossRef CAS PubMed.
- K. Wang, W. Shi, J. Jia, S. Chen and H. Ma, Talanta, 2009, 77, 1795–1799 CrossRef CAS PubMed.
- T. Gunnlaugsson, P. E. Kruger, T. C. Lee, R. Parkesh, F. M. Pfeffer and G. M. Hussey, Tetrahedron Lett., 2003, 44, 6575–6578 CrossRef CAS.
- M. Lazzarotto, F. F. Nachtigall, E. Schnitzler and E. E. Castellano, Thermochim. Acta, 2005, 429, 111–117 CrossRef CAS.
- S. Elçin and H. Deligöz, Tetrahedron, 2013, 69, 6832–6838 CrossRef.
- S. Elçin, G. K. Çılgı, A. Bayrakdar and H. Deligöz, Spectrochim. Acta, Part A, 2015, 142, 178–187 CrossRef PubMed.
- R. W. Berg, Appl. Spectrosc. Rev., 2015, 50, 193–239 CrossRef CAS.
- P. Singh, N. P. Singha and R. A. Yadavb, J. Chem. Pharm. Res., 2010, 2, 656–681 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. M. Gill, B. G. Johnson, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1992, 197, 499–505 CrossRef CAS.
- R. G. Parr and W. Yang, Density-functional theory of atoms and molecules, Oxford University Press, 1989 Search PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- R. Reslan, K. Lopata, C. Arntsen, N. Govind and D. Neuhauser, J. Chem. Phys., 2012, 137, 22A502 CrossRef PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedure, full structural characterization data, and absorption/fluorescence spectral data for quinoxalinium salts 1–5 are provided. See DOI: 10.1039/c6ra14134j |
| ‡ Shared first author (contributed equally). |
| This journal is © The Royal Society of Chemistry 2016 |