
Effective tumor-targeted delivery of etoposide using chitosan nanoparticles conjugated with folic acid and sulfobetaine methacrylate†
Abstract
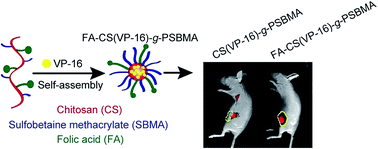
We demonstrated chitosan (CS)-based biocompatible nanoparticles coated with folic acid (FA) and poly(sulfobetaine methacrylate) (PSBMA) as an effective tumor-specific drug delivery system. The graft copolymer FA–CS-g-PSBMA could self-assemble into nanoparticles in an aqueous phase and maintain a spherical shape. Etoposide (VP-16), a widely-used chemotherapy drug with poor water solubility, could be incorporated into the inner core of hydrophobic CS to form FA–CS(VP-16)-g-PSBMA nanoparticles. The synthesis of the nanocarrier was verified by using zeta potential analysis, 1H nuclear magnetic resonance and Fourier transform infrared spectra. Next, both in vitro and in vivo experiments were performed to evaluate the release behavior, cellular uptake, cytotoxicity, biodistribution and therapeutic efficacy of the nanoparticles. Our results showed FA–CS(VP-16)-g-PSBMA nanoparticles released VP-16 more effectively in acidic phosphate-buffered saline than that under neutral conditions, and could be effectively internalized into HeLa cells. Compared to the nanoparticles without FA, FA–CS(VP-16)-g-PSBMA nanoparticles exhibited a more significant inhibitory effect on HeLa cell viability in vitro. When HeLa tumor-bearing mice were intravenously administrated with fluorescence-labelled nanoparticles, FA-conjugated nanoparticles accumulated more rapidly at the tumor site. Furthermore, FA–CS(VP-16)-g-PSBMA nanoparticles demonstrated more superior therapeutic efficacy than VP-16. These results suggest that FA–CS-g-PSBMA nanoparticles represent a promising nanocarrier for anti-tumor drug delivery.
 Please wait while we load your content...
Please wait while we load your content...

