
Surface carbon species formation from ethylene decomposition on Pd(100): a first-principles-based kinetic Monte Carlo study
Abstract
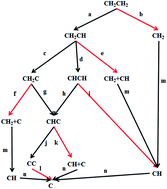
Based on the activation barriers and reaction energies from periodic density functional calculations, we conducted kinetic Monte Carlo (kMC) simulations of surface carbon species formation from ethylene decomposition on a Pd(100) surface. A comprehensive reaction network of ethylene decomposition involving such intermediates as CH2CH, CHCH, CH2C, CHC, CC, CH2 and CH was proposed. Our kMC simulations show that the most probable pathway of ethylene decomposition on Pd(100) is CH2CH2 → CH2CH → CH2C → CHC → CC, among which the dehydrogenation of CH2CH2 to CH2CH is the rate-limiting step with the activation barrier of 1.51 eV, followed by CH2CH2 → CH2CH → CHCH → CHC → CC, whose rate-limiting step is the dehydrogenation of CH2CH to CHCH with the activation barrier of 1.59 eV. The two most probable pathways produce a carbon dimer as the final product, since the activation barrier of the C–C bond cleavage reaction is so high (2.32 eV) that it is almost impossible for it to occur before the metal surface is totally poisoned by surface carbon species. Another three feasible pathways are: (i) CH2CH2 → CH2CH → CHCH → CH → C, (ii) CH2CH2 → CH2CH → CHCH → CHC → CH + C → C and (iii) CH2CH2 → CH2CH → CH2C → CHC → CH + C → C, whose final products contain surface carbon monomers. And the reactions involving C–C bond cleavage are the rate-limiting step of the three pathways. Simple as the reaction network of ethylene decomposition looks, it is still difficult to analyze the decomposition mechanism merely according to the activation barriers from DFT calculations. Our work here demonstrates that kMC simulations can nicely tackle the problem on competitive reaction pathways, each of which involves some reactions with relatively low activation barriers (e.g. the dehydrogenation reactions involved in ethylene decomposition) and some other reactions with relatively high activation barriers (e.g. the C–C bond cleavage reactions involved in ethylene decomposition).
 Please wait while we load your content...
Please wait while we load your content...

