
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic enantioselective oxa-hetero-Diels–Alder reactions of enones with aryl trifluoromethyl ketones†
Dongxin Zhang and
Fujie Tanaka*
Chemistry and Chemical Bioengineering Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna, Okinawa 904-0495, Japan. E-mail: ftanaka@oist.jp
First published on 21st June 2016
Abstract
The development of oxa-hetero-Diels–Alder reactions of enones with aryl trifluoromethyl ketones to afford tetrahydropyranones bearing trifluoromethyl-substituted tetrasubstituted carbon centers is reported. The reactions were catalyzed by an amine-based catalyst system and afforded the products with er values up to 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.
:3.
Tetrahydropyranones and tetrahydropyrans are important structures found in bioactive natural products and pharmaceutical leads.1,2 Incorporation of the trifluoromethyl group has been shown to favour bioactivity,3 therefore concise routes to tetrahydropyranone and tetrahydropyran derivatives bearing a trifluoromethyl group are of interest. To synthesize functionalized tetrahydropyranones, we have recently developed enantioselective oxa-hetero-Diels–Alder reactions of enones with isatins that are catalyzed by amine-based catalyst systems.2 In the reactions, enamines of enones are formed in situ, and the enamines act as dienes of the [4 + 2] cycloaddition resulting in the formation of the tetrahydropyranones under mild conditions.2 Based on these studies, we reasoned that oxa-hetero-Diels–Alder reactions of enones with trifluoromethyl ketones would provide access to trifluoromethyl-substituted tetrahydropyran derivatives. However, direct use of enones as diene precursors to form tetrahydropyranones is still a challenge; reported reactions of enones with ketones or aldehydes often give aldol products as the main product or as a significant by-product.4 That is, formation of oxa-hetero-Diels–Alder reaction product is not promised in the reactions of enones with ketones or aldehydes as dienophiles either in racemic or highly enantioselective versions.2,5 Here, we report enantioselective oxa-hetero-Diels–Alder reactions of enones with aryl trifluoromethyl ketones that afford trifluoromethyl-substituted tetrahydropyranones (Scheme 1).
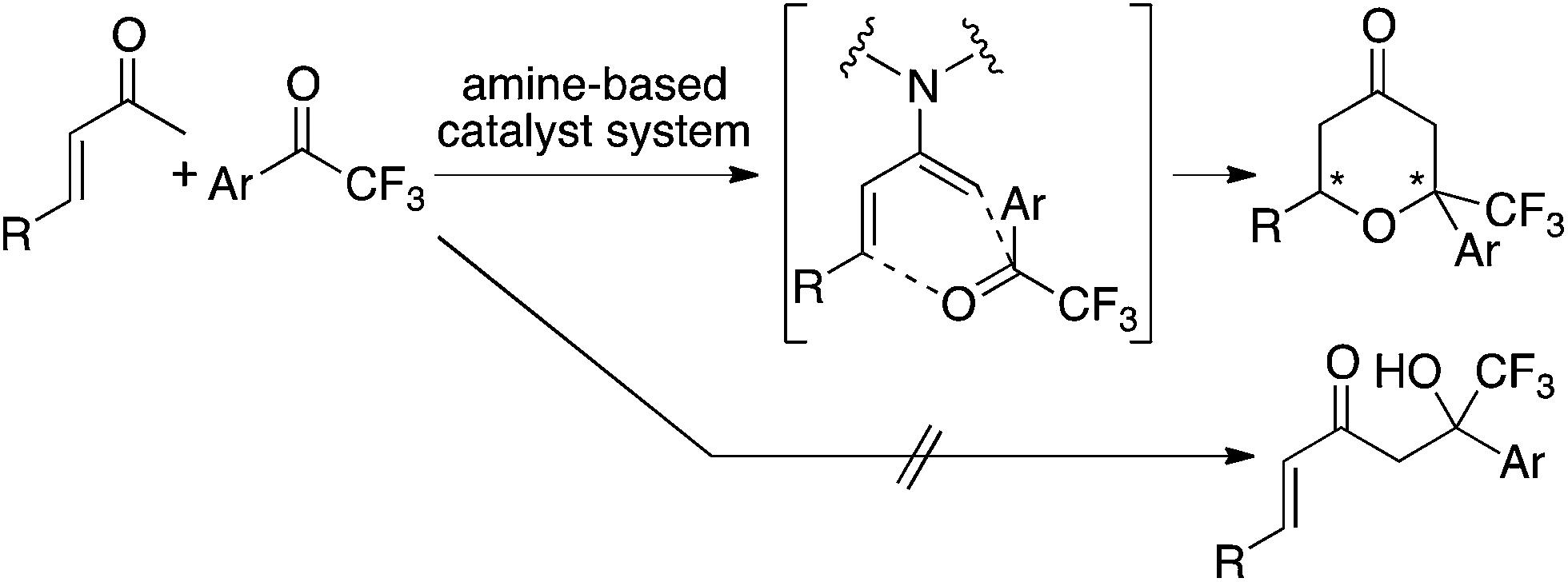 | ||
| Scheme 1 The oxa-hetero-Diels–Alder reactions of enones with aryl trifluoromethyl ketones catalyzed by amine-based catalyst systems to afford trifluoromethyl-substituted tetrahydropyranones. | ||
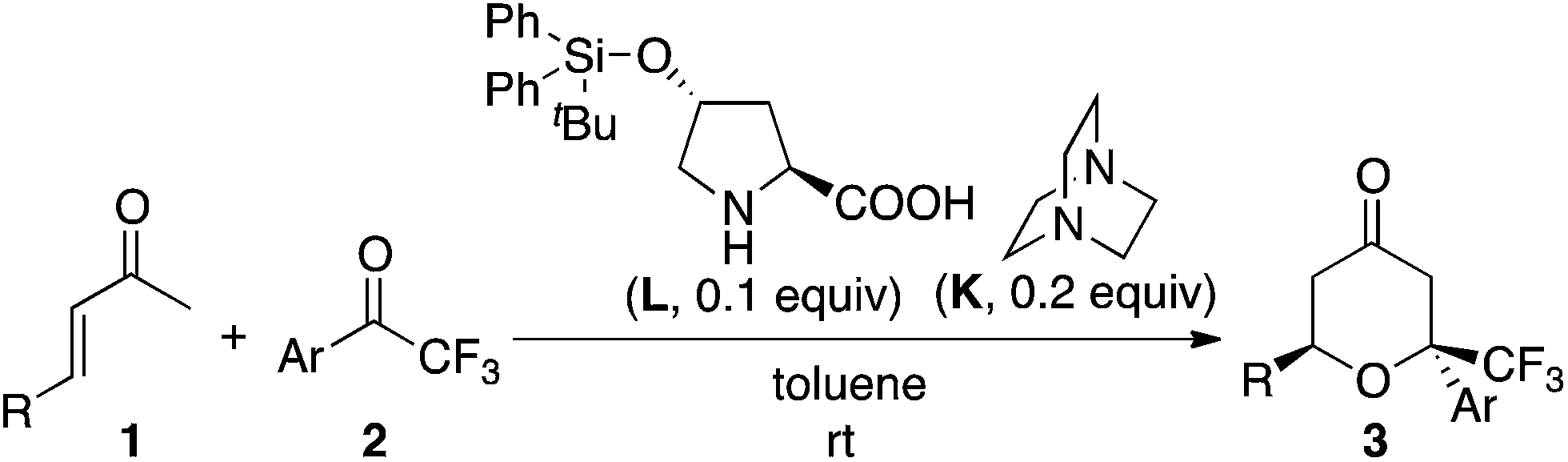
First, we screened catalyst systems for the reaction of enone 1a with ketone 2a to form trifluoromethyl-substituted tetrahydropyranone product 3aa (3aa-1 and/or 3aa-2). Selected results are shown in Table 1. Previously reported catalyst systems (such as A–B, A–B–C, and D–B) for the reactions of enones with isatins to afford tetrahydropyranones in high enantioselectivity2 did not work efficiently for the reaction with ketone 2a; the use of these catalysts significantly generated aldol product 4aa with oxa-hetero-Diels–Alder product 3aa (Table 1, entries 1–3). The best results for the formation of 3aa with high enantioselectivity (er 97:3 for 3aa-2) were obtained when the reaction was performed in the presence of proline-derived catalyst L and DABCO (K) in toluene at rt (25 °C) (Table 1, entries 11 and 12). The reaction using less loading of L (0.1 equiv.) with K (0.2 equiv.) gave essentially the same results as the reaction using L (0.2 equiv.) and K (0.2 equiv.) (Table 1, entry 12 versus entry 11). The major diastereomer (i.e., 3aa-2) obtained under the catalysis by L–K differed from that obtained under the catalysis by A–B (Table 1, entries 11 and 12 versus entry 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst system | Time (h) | 3aa:4aab |
drb 3aa-1:3aa-2 |
erc 3aa-1/3aa-2 |
| a Reaction was performed by using enone 1a (0.5 mmol) and aryl trifluoromethyl ketone 2a (0.1 mmol) in the presence of the indicated catalyst system in toluene (0.2 mL) at 25 °C until 2a was consumed except where indicated. The relative stereochemistry of 3aa-1 and 3aa-2 was determined to be as shown; the absolute stereochemistry of 3aa-1 and 3aa-2 is tentative; see ESI.b Determined by 1H NMR analysis of the crude mixture.c Determined by HPLC analysis. ND = not determined.d Conversion <20%.e Reaction in DMF. | |||||
| 1 | A (0.2 equiv.)–B (0.4 equiv.) | 24 | 62:38 |
5.0:1 |
85:15/20:80 |
| 2 | A (0.2 equiv.)–B (0.4 equiv.)–C (0.4 equiv.) | 36 | 71:29 |
2.5:1 |
ND/ND |
| 3 | D (0.2 equiv.)–B (0.4 equiv.) | 12 | 67:33 |
3.1:1 |
ND/ND |
| 4 | E (0.2 equiv.)–F (0.4 equiv.) | 24 | 95:5 |
2.0:1 |
18:82/1:1 |
| 5 | G (0.2 equiv.)–F (0.4 equiv.) | 24 | >95:5 |
1.7:1 |
68:32/ND |
| 6d | H (0.2 equiv.) | 48d | — | — | — |
| 7e | H (0.2 equiv.) | 24 | >95:5 |
1.6:1 |
ND/85:15 |
| 8 | H (0.2 equiv.)–I (0.2 equiv.) | 36 | >95:5 |
1.3:1 |
ND/91:9 |
| 9 | H (0.2 equiv.)–J (0.2 equiv.) | 30 | >95:5 |
1:2.3 |
ND/91:9 |
| 10 | H (0.2 equiv.)–K (0.2 equiv.) | 36 | >95:5 |
1:1.2 |
ND/95:5 |
| 11 | L (0.2 equiv.)–K (0.2 equiv.) | 24 | >95:5 |
1:1.9 |
1:1/97:3 |
| 12 | L (0.1 equiv.)–K (0.2 equiv.) | 24 | >95:5 |
1:1.9 |
1:1/97:3 |
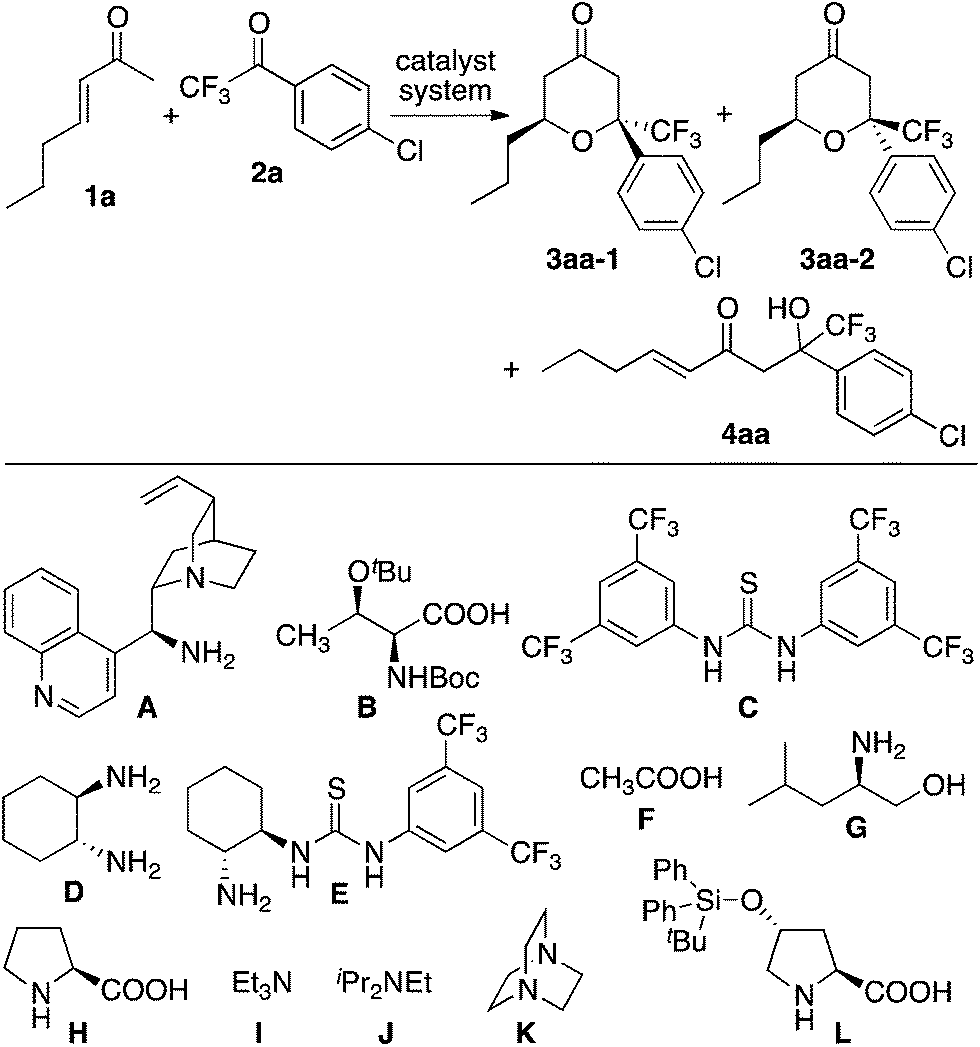
Next, using the best catalyst system identified [i.e., L (0.1 equiv.)–K (0.2 equiv.)], reactions of various enones and aryl trifluoromethyl ketones were performed (Table 2). In all cases, trifluoromethyl-substituted tetrahydropyranones were obtained with high enantioselectivities for the major diastereomer products, and tetrasubstituted carbon centers were concisely constructed (Table 2). The reactions of phenyl trifluoromethyl ketones bearing electron-withdrawing substituents on the phenyl group (such as the formation of 3ad) were faster than the reactions of those bearing electron-donating groups (such as the formation of 3af). In all cases shown in Table 2, the formation of the aldol product was negligible (3:4 were >95:5 or 95:5).
|
|---|
| a Reaction conditions: enone 1 (1.0 mmol) and aryl trifluoromethyl ketone 2 (0.2 mmol) in the presence of proline derivative L (0.02 mmol) and DABCO (K, 0.04 mmol) in toluene (0.4 mL) at 25 °C. The isolated yields of 3 (combined for both the diastereomers) are shown except where noted. The dr was determined by 1H NMR analysis before purification. The er of the major diastereomer was determined by HPLC analysis. The ratio 3:4 (4 = aldol product) was determined by 1H NMR analysis before purification: >95:5 for the formation of 3aa, 3ab, 3ac, 3ad, 3ae, 3af, 3ag, 3ah, 3bb, 3bc, and 3bd; 95:5 for the formation of 3ba.b Ketone 2 was not consumed.c Data of 1 mmol-scale reaction; isolated yield of the major isomer, the dr of the major diastereomer after purification. |
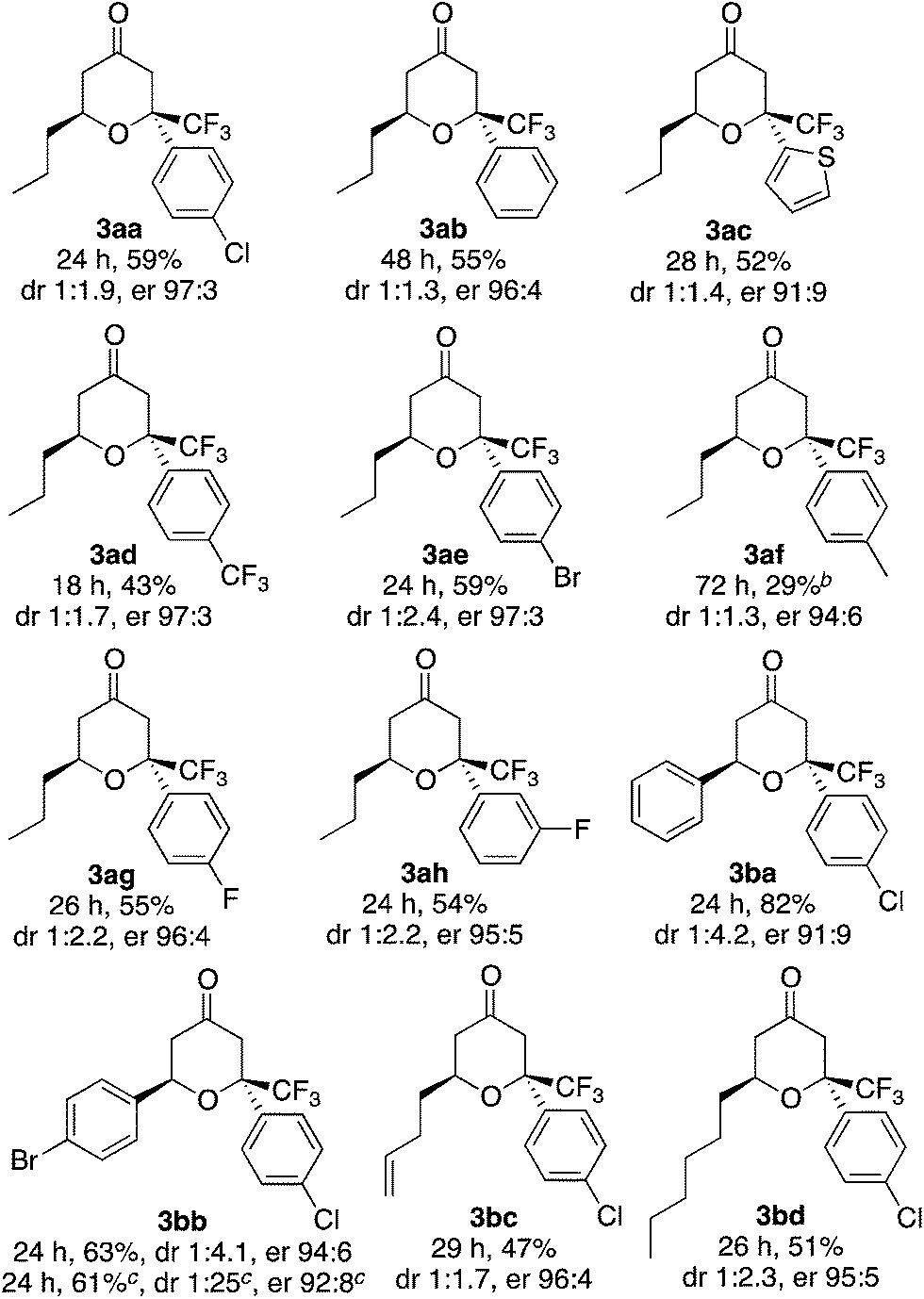 |
The catalyst system was useful for the reactions of β-alkyl substituted enones and also β-aryl substituted enones to afford the hetero-Diels–Alder reaction products with high enantioselectivities for the major product diastereomers. This is significant because previously reported conditions for the hetero-Diels–Alder reactions of β-alkyl substituted enones often do not work for the β-aryl substituted enones.2,5c
Further, the reaction using the L–K catalyst system was easily scaled up: a 1.0 mmol-reaction to form 3bb gave the major isomer, 3bb-2, as a single diastereomer (purity >95%) in 61% yield with er 92:8.
When a mixture of 3aa and 4aa (racemic, 3aa/4aa = 2.5:1, 3aa-1:3aa-2 = 3:1) was treated under the hetero-Diels–Alder reaction conditions with the L–K catalyst system, no decomposition of the compounds and no changes in the ratios were detected. This indicates that product 3aa is stable under the L–K catalyst system and that aldol 4aa is not converted to 3aa in the presence of this catalyst system. Thus, the formation of 3aa under the L–K catalyst system is likely a kinetically controlled [4 + 2] cycloaddition reaction of in situ-generated enamine of enone 1aa with ketone 2aa.
To demonstrate the use of the hetero-Diels–Alder reactions, the product tetrahydropyranones were transformed into tetrahydropyran derivatives (Scheme 2). Oxime formation, reductive amination, and allylation gave the corresponding products 5–8. The trifluoromethyl-substituted tetrahydropyranones and tetrahydropyran derivatives that can be synthesized by the methods described here may be useful in the search for biofunctional molecules.
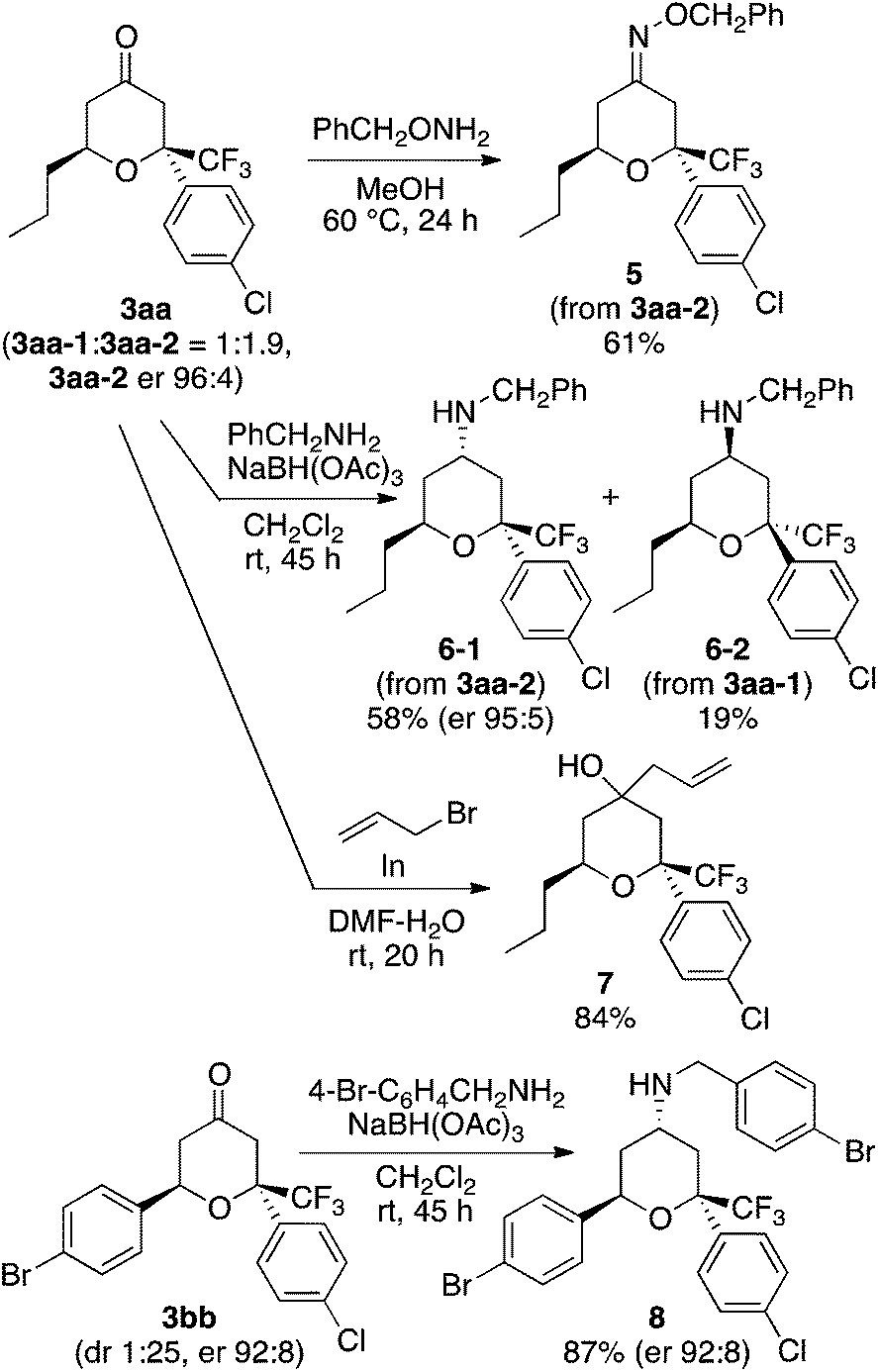 | ||
| Scheme 2 Transformations of the hetero-Diels–Alder products. | ||
In conclusion, we have developed an organocatalytic enantioselective oxa-hetero-Diels–Alder reaction of enones with aryl trifluoromethyl ketones that afford trifluoromethyl-substituted tetrahydropyranones, which uses novel amine-based catalyst systems. Tetrasubstituted carbon centers bearing a trifluoromethyl group were concisely constructed with the formation of the tetrahydropyranone ring. We have also demonstrated that the hetero-Diels–Alder products can be transformed further to various trifluoromethyl-substituted tetrahydropyran derivatives.
Acknowledgements
We thank Dr Michael Chandro Roy, Research Support Division, Okinawa Institute of Science and Technology Graduate University for mass analyses. This study was supported by the Okinawa Institute of Science and Technology Graduate University and by the MEXT (Japan) Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” (No. 26105757).Notes and references
- (a) G. Dossetter, T. F. Jamison and E. N. Jacobsen, Angew. Chem., Int. Ed., 1999, 38, 2398 CrossRef; (b) Y. Yamashita, S. Saito, H. Ishitani and S. Kobayashi, J. Am. Chem. Soc., 2003, 125, 3793 CrossRef CAS PubMed; (c) M. Anada, T. Washio, N. Shimada, S. Kitagaki, M. Nakajima, M. Shiro and S. Hashimoto, Angew. Chem., Int. Ed., 2004, 43, 2665 CrossRef CAS; (d) A. K. Unni, N. Takenaka, H. Yamamoto and V. H. Rawal, J. Am. Chem. Soc., 2005, 127, 1336 CrossRef CAS PubMed; (e) S. Rajaram and M. S. Sigman, Org. Lett., 2005, 7, 5473 CrossRef CAS PubMed; (f) N. Momiyama, H. Tabuse and M. Terada, J. Am. Chem. Soc., 2009, 131, 12882 CrossRef CAS PubMed; (g) J. Guin, C. Rabalakos and B. List, Angew. Chem., Int. Ed., 2012, 51, 8859 CrossRef CAS PubMed; (h) T. Voigt, C. Gerding-Reimer, T. T. N. Tran, S. Bergmann, H. Lachance, B. Scholermann, A. Brockmeyer, P. Janning, S. Ziegler and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 410 CrossRef CAS PubMed; (i) N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323 RSC; (j) G. C. Tay, C. H. Huang and S. D. Rychnovsky, J. Org. Chem., 2014, 79, 8733 CrossRef CAS PubMed.
- (a) H.-L. Cui and F. Tanaka, Chem.–Eur. J., 2013, 19, 6213 CrossRef CAS PubMed; (b) H.-L. Cui, P. V. Chouthaiwale, F. Yin and F. Tanaka, Asian J. Org. Chem., 2016, 5, 153 CrossRef CAS; (c) H.-L. Cui, P. V. Chouthaiwale, F. Yin and F. Tanaka, Org. Biomol. Chem., 2016, 14, 1777 RSC , and references cited therein.
- (a) K. Mueller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) P. Wang, L.-W. Feng, L. Wang, J.-F. Li, S. Liao and Y. Tang, J. Am. Chem. Soc., 2015, 137, 4626 CrossRef CAS PubMed; (d) S. Kawamura, H. Egami and M. Sodeoka, J. Am. Chem. Soc., 2015, 137, 4865 CrossRef CAS PubMed; (e) D. Zhang and F. Tanaka, Adv. Synth. Catal., 2015, 357, 3458 CrossRef.
- (a) B. M. Trost, S. Shin and J. A. Sclafani, J. Am. Chem. Soc., 2005, 127, 8602 CrossRef CAS PubMed; (b) Q. Guo, M. Bhanushali and C.-G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 9460 CrossRef CAS PubMed; (c) G. Pousse, F. Le Cavelier, L. Humphreys, J. Rouden and J. Blanchet, Org. Lett., 2010, 12, 3582 CrossRef CAS PubMed; (d) G.-G. Liu, H. Zhao, Y.-B. Lan, B. Wu, X.-F. Huang, J. Chen, J.-C. Tao and X.-W. Wang, Tetrahedron, 2012, 68, 3843 CrossRef CAS; (e) T. Yan, X. Wang, H. Sun, J. Liu and Y. Xie, Molecules, 2013, 18, 14505 CrossRef PubMed; (f) C. Baker-Glenn, N. Hodnett, M. Reiter, S. Ropp, R. Anclif and V. Gouverneur, J. Am. Chem. Soc., 2005, 127, 1481 CrossRef CAS PubMed; (g) S. Abbaraju and J. C.-G. Zhao, Adv. Synth. Catal., 2014, 356, 237 CrossRef CAS PubMed.
- (a) L.-Q. Lu, X.-N. Xing, X.-F. Wang, Z.-H. Ming, H.-M. Wang and W.-J. Xiao, Tetrahedron Lett., 2008, 49, 1631 CrossRef CAS; (b) M. Mojzesova, M. Meciarova, M. Marti and R. Sebesta, New J. Chem., 2015, 39, 2573 RSC; (c) Y.-J. Lin, L.-N. Du, T.-R. Kang, Q.-Z. Liu, Z.-Q. Chen and L. He, Chem.–Eur. J., 2015, 21, 11773 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of compounds, 1H and 13C NMR spectra, and HPLC charts. See DOI: 10.1039/c6ra13859d |
| This journal is © The Royal Society of Chemistry 2016 |