
Abstract
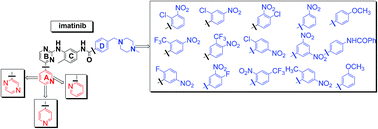
We revisited the classical synthetic procedure for imatinib synthesis providing an improved and optimized approach in the preparation of a series of new imatinib analogues. The proposed methodology effectively overcomes certain problematic steps, saves time and labor, provides a very high yield and purity and has the potential to be used for the synthesis of many analogues. The formation of the desired guanidine salt 4, one of the key steps to the imatinib synthesis, was proceeded almost quantitatively by the reaction of the hydrochloride of the suitable aniline 3 with excess of molten cyanamide, without any solvent. Pure arylamine intermediates 6a–d were obtained quantitatively in a short reaction time after reduction of the nitro group of the intermediate pyrimidines 5a–d with hydrogen over the Adam's catalyst. In addition, the application of this optimized approach can be extended in the synthesis of nilotinib and its analogues intermediates.
 Please wait while we load your content...
Please wait while we load your content...

