
Fe, S co-doped anatase TiO2 nanotubes as anodes with improved electrochemical performance for lithium ion batteries†
Abstract
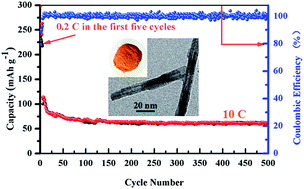
Fe, S co-doped anatase TiO2 nanotubes (FSTNTs), where all of the Fe atoms and most of the S atoms substitute for lattice Ti with only few sulfur atoms replacing with lattice O, are successfully prepared by a facile sol–gel process and subsequent chemical method. The as-prepared products are straight nanotubes with inner diameters of 5–8 nm and outer diameters of 10–30 nm. Owing to the enhancement of both thermodynamic and kinetic properties by the Fe, S dopant pair, the doping level and electrical conductivity of FSTNTs are greatly increased. When firstly used as anodes for lithium ion batteries, the FSTNTs electrodes exhibit excellent cycling stabilities (around 140 mA h g−1 at 4C after 50 cycles and 61.4 mA h g−1 at 10C after 500 cycles) and remarkable rate performances (88.4 mA h g−1 at 10C and 51.5 mA h g−1 at 20C).
 Please wait while we load your content...
Please wait while we load your content...

