
Ionic liquid-based uranium(vi) extraction with malonamide extractant: cation exchange vs. neutral extraction†
Abstract
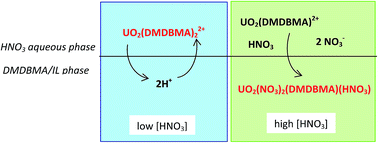
We present new insights into the extraction of uranium(VI) from a nitric acid aqueous phase into 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ionic liquid ([C4mim][Tf2N]) using a malonamide extractant, namely N,N′-dimethyl-N,N′-dibutylmalonamide (DMDBMA). UV-vis absorption spectrophotometry and extended X-ray absorption fine structure (EXAFS) experiments have been carried out on the extracted phases and new extraction data were used in order to model the mechanism lying behind the U(VI) extraction. We show that two different uranyl species are involved, as a function of the aqueous nitric acid concentration: the cation UO2(DMDBMA)x2+ (2 ≤ x ≤ 3) at low acid concentration, and the neutral UO2(NO3)2(DMDBMA) at high acid concentration. The former is extracted by exchange with 2 protons, while the latter is co-extracted with a HNO3 molecule. We show that the uranium extraction is performed without the direct help of IL ions, although the latter pollute noticeably the aqueous phase.
 Please wait while we load your content...
Please wait while we load your content...

