
Magnetic solid phase extraction of polycyclic aromatic hydrocarbons and chlorophenols based on cyano-ionic liquid functionalized magnetic nanoparticles and their determination by HPLC-DAD
Abstract
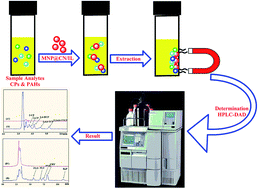
A magnetic solid-phase extraction (MSPE) procedure on the newly synthesized cyano-ionic liquid functionalized magnetic nanoparticles (MNP@CN/IL) was established and validated for the separation/preconcentration of polycyclic aromatic hydrocarbons (PAHs) and chlorophenols (CPs) from water samples prior to their HPLC-DAD determination. The new magnetic (MNP@CN/IL) adsorbent was characterized by Fourier transform infrared (FT-IR) spectroscopy, X-ray diffraction (XRD), elemental analysis (CHNS), energy-dispersive X-ray spectroscopy (EDX), vibrating sample magnetometry (VSM), thermal gravimetric analysis (TGA) and transmission electron microscopy (TEM). The proposed method showed preconcentrations of 200 and 100 with low limits of detection (LOD = 0.40–0.59 and 0.35–0.67 μg L−1) with linearity ranges from 0.1–100 and 3–100 μg L−1 for PAHs and CPs, respectively. The extraction recoveries from environmental samples (leachate and sludge from a landfill site) ranged from 89.50–110.2% and 80.67–112.7% for PAHs and CPs, respectively, with satisfactory precision (% RSD less than 5%). The combination of cyano group and ionic liquid on the surface of the MNPs enhanced the extraction efficiency which might be due to π–π interactions as well as electrostatic interactions.
 Please wait while we load your content...
Please wait while we load your content...

