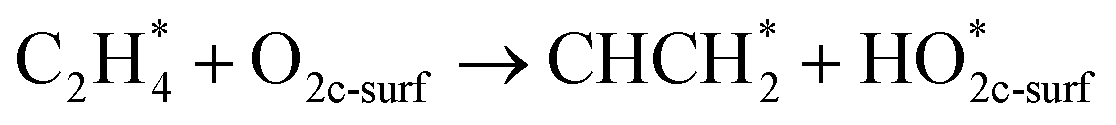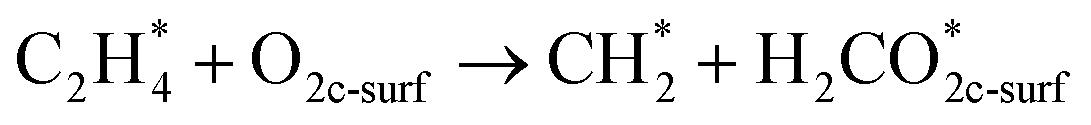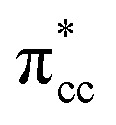DOI:
10.1039/C6RA13633H
(Paper)
RSC Adv., 2016,
6, 92241-92251
Enhanced molecular adsorption of ethylene on reduced anatase TiO2 (001): role of surface O-vacancies†
Received
26th May 2016
, Accepted 12th September 2016
First published on 12th September 2016
Abstract
A density functional theory (DFT)-based study of ethylene (C2H4) adsorption on a reduced anatase titanium dioxide (TiO2) (001) surface, i.e., with a surface oxygen vacancy (Ovac), is presented. It was found that C2H4 preferably adsorbs on the Ovac-site. The excess electrons originating from the removed oxygen weaken the C2H4 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond by filling the lowest unoccupied molecular orbital (LUMO) or
C double bond by filling the lowest unoccupied molecular orbital (LUMO) or  of C2H4, and simultaneously enhance the binding of C2H4 to the reduced anatase TiO2 (001). The bonding between the two C2H4 C atoms and the two Ti atoms nearest to the Ovac-site produces two σ-type bonds, leading to the emergence of a new localized mid-gap state. This hybrid
of C2H4, and simultaneously enhance the binding of C2H4 to the reduced anatase TiO2 (001). The bonding between the two C2H4 C atoms and the two Ti atoms nearest to the Ovac-site produces two σ-type bonds, leading to the emergence of a new localized mid-gap state. This hybrid 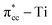 3d defect state can account for the ∼0.7 eV decrease in the band-gap observed in previous optical measurements of carbon-coated TiO2, formed after TiO2 exposure to C2H4 gas (Mater. Lett. 108, 2013, 134). Subsequent calculations on two initial decomposition pathways of C2H4 adsorbed on the Ovac-site show that the C–H bond and C–C bond cleaving require high activation barriers where the C–H bond cleaving is slightly easier compared to the C–C bond cleaving, 2.94 eV and 3.01 eV, respectively, (as calculated using DFT-D2+Ud = 3 eV) and the final states of the two initial decomposition pathways show similar endothermic characteristics. This finding indicates that the surface Ovac-sites tend to favor the formation of molecularly adsorbed Ti-bound sp3-C2H4 (–Ti–CH2CH2–Ti–) as compared to the dissociative adsorption case.
3d defect state can account for the ∼0.7 eV decrease in the band-gap observed in previous optical measurements of carbon-coated TiO2, formed after TiO2 exposure to C2H4 gas (Mater. Lett. 108, 2013, 134). Subsequent calculations on two initial decomposition pathways of C2H4 adsorbed on the Ovac-site show that the C–H bond and C–C bond cleaving require high activation barriers where the C–H bond cleaving is slightly easier compared to the C–C bond cleaving, 2.94 eV and 3.01 eV, respectively, (as calculated using DFT-D2+Ud = 3 eV) and the final states of the two initial decomposition pathways show similar endothermic characteristics. This finding indicates that the surface Ovac-sites tend to favor the formation of molecularly adsorbed Ti-bound sp3-C2H4 (–Ti–CH2CH2–Ti–) as compared to the dissociative adsorption case.
1 Introduction
There has been growing interest in the study of transition metal oxide–hydrocarbon molecule interactions in the past few years. This results from the wide applicability of the interaction in various fields. Some related applications include the decomposition of organic pollutants derived from volatile organic compounds,1–4 petroleum chemical processes,5,6 preservation of fresh foods,7 and sensitizing oxide surfaces with hydrocarbon-derived substances for solar cell devices.8–10
In this paper particular attention is given to the ethylene–titanium dioxide (C2H4–TiO2) interaction. Previous studies of the photocatalytic decomposition of C2H4 using TiO2 found that in the absence of light irradiation, C2H4 did not adsorb nor dissociate with TiO2. Experimental measurement shows no gas pressure change occurred even after long exposure of TiO2 to C2H4. More recently Fourier-transform-infrared measurements on low temperature C2H4 adsorption using a Degussa P25-type of TiO2 showed that C2H4 interacts weakly with TiO2. The C2H4 vibrational frequencies did not undergo significant modification and C2H4 retains its planar configuration upon adsorption.11–14 These results agree with theoretical calculations on C2H4 adsorption on a pristine anatase TiO2 (001) surface.15 It was found that C2H4 adsorbs on anatase TiO2 (001) via a π-complex interaction with the unsaturated five-coordinated Ti (Ti5c). Furthermore, it was found that the dispersion (van der Waals) interaction further enhances the binding of C2H4.
A number of further studies on C2H4–TiO2 have since been conducted, most of them focusing on C2H4 decomposition using TiO2 as a photocatalyst.16–20 Some recent experimental work have tried to modify TiO2 surfaces with carbon by using a hydrocarbon such as C2H4 and acetylene as carbon sources.21,22 Some other researchers have also have attempted to grow carbonaceous structures on TiO2 for solar cell device application.23,24 Indeed, these studies have given many valuable insights to understand the nature of C2H4–TiO2 interaction. However, not much is known regarding the role of intrinsic defects to C2H4 interaction with TiO2. Intrinsic defects, in particular oxygen vacancy (Ovac), are expected to have important roles in determining the physics and chemistry of transition metal oxide surfaces. The excess electrons originating from the Ovac often become a critical factor that governs the interaction of oxide surfaces with various kind of electron-acceptor molecules.25–29 Therefore, it is necessary to study further how C2H4 adsorption – which can be regarded as an olefin prototype – behaves when an Ovac exists in TiO2.
In the present work, TiO2 anatase was chosen because of its relatively higher catalytic activity compared to its rutile counterpart.30–32 For the surface facet, research was concentrated on the bulk-terminated (1 × 1) anatase TiO2 (001) because of its superior catalytic activity, in particular when coupled with the presence of an Ovac.33,34 Indeed, bulk-terminated anatase TiO2 (001) is well known for its highly unstable characteristics, and thus, it usually undergoes surface reconstruction (the so-called (1 × 4)-reconstructed) to compensate for this instability. However, recent experimental work has succeeded in synthesizing anatase nanoparticles with the dominant bulk-terminated (001) facet. This is achieved by using a particular non-metallic atom (typically fluorine), which acts as a shape-controlling agent. Furthermore, this shape-controlling agent can be cleaned using heat treatment to produce a fluorine free surface without changing the basic crystal structure and morphology.35–37 Based on these useful results, an attempt has been made to further explore the interaction between C2H4 and reduced anatase TiO2 (001) surface. In this study, it will be shown that the presence of the Ovac promotes the formation of molecularly adsorbed C2H4. Furthermore, it will also be shown how the coupling between the C2H4-reduced anatase TiO2 (001) can possibly extend the photoabsorption of TiO2 to the visible light range. A similar study has been performed on the more stable (101) facet, as well. There are some differences in the energetics and configurations of the adsorbed C2H4, in particular, on the reduced anatase TiO2 (101) surface with that on the reduced (001) surface. This originates mainly from the difference of the preferable site of the Ovac. The (101) surface favors the formation of the Ovac in the subsurface, which is different to the (001) surface which favors the formation of the Ovac on its outer most layer (this will be explained in Section 3.1). However, it is planned to publish the results in a future work to ensure a more rigorous and concise report.
2 Computational details
Spin-polarized density functional theory (DFT)-based calculations were carried out within the projector augmented-wave (PAW) method, as implemented in the Vienna Ab-initio Simulation Package (VASP) code.38–40 The Generalized Gradient Approximation (GGA) as formulated by Perdew–Burke–Ernzerhof (PBE) was used to describe the exchange interaction and electronic correlation.41,42 A plane-wave basis set was used with a kinetic cut-off energy of 520 eV. The geometric optimizations were carried out with residual forces less than 0.01 eV Å−1. The calculated bulk lattice parameters for anatase TiO2 are a = b = 3.83 Å and c = 9.66 Å, and these results are in good agreement with the results from previous work.43,44
Approximately 16 Å of vacuum space was utilized and included a dipole correction to eliminate the artificial interaction between the periodically repeated slabs. A c(2 × 2) unit cell, was utilized containing four unsaturated Ti5c atoms, each bonding to two-raised 2-coordinated (O2c) and two-lowered 3-coordinated (O3c) oxygen atoms along the [100] and [010] directions, respectively. Brillouin zone integration was sampled with 6 × 6 × 1 k-points. Each slab consists of four O–Ti–O layers. This amount of layers is sufficient to produces good convergence of the (001) surface structure32,45 and has been widely used in previous studies.46–48 Two upper O–Ti–O layers of the slab were allowed to relax during geometry optimization, keeping the other remaining O–Ti–O layers at the bottom side fixed to its initial bulk position. The adsorption energy is defined as:
| | |
Eads = Eadsorbate/surface − (Eadsorbate + Esurface)
| (1) |
where
Eadsorbate is the energy of isolated C
2H
4 in the vacuum.
Esurface is the total energy of the anatase (001) surface and
Eadsorbate/surface is the total energy of the surface together with the adsorbed molecule. A long range dispersion interaction [or van der Waals (vdW) interaction] was included,
49 i.e.| |
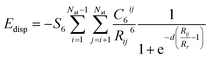 | (2) |
The damping factor, S6, is 0.75 as the GGA-PBE was utilized in all calculations. The dispersion coefficient (C6) and the vdW radius (R0) for O, C, and H are 0.7 J nm6 mol−1, 1.75 J nm6 mol−1, 0.14 J nm6 mol−1 and 1.342 Å, 1.452 Å, 1.001 Å, respectively.49 For Ti, C6 and R0 are set to be 10.8 J nm6 mol−1 and 1.562 Å, following previous theoretical studies.50
Previous theoretical studies on TiO2 with an Ovac have shown that a common GGA functional failed to describe the experimentally observed localized defect states, because of inherent spurious self-interaction within the DFT formalism. This results in unphysical delocalization of the electronic states.51–53 The utilization of DFT-D2+Ud, however, could overcome this problem. This method is implemented by introducing the on-site Coulomb interaction in the localized d-orbital and exchange interactions, by adding an effective Hubbard-U parameter to better describe the repulsion between the electrons on the same orbital.54,55 Here, Ud = 3 eV was mainly used to determine the bonding mechanism and energetics of the initial decomposition reactions of C2H4 on reduced anatase TiO2 (001). Indeed, the chosen Ud is not sufficient to fully open the band-gap of pristine anatase TiO2 to its experimentally measured value (∼2.4 eV versus ∼3.2 eV). However, there are some considerations that have been taken into account as to why the present Ud was chosen: (1) the present choice of Ud properly describes the energy level of the reduced anatase TiO2 (001) defect state with respect to the conduction band minimum (this will be elaborated further in the next section); (2) it gives a reasonable estimation for the reaction energies of TiO2 with other molecules [e.g., hydrogen (H2)];56 (3) if the Ud-parameter (typically >5 eV) is too high it may open the band-gap more, but simultaneously it may also result in the wrong description of the adsorbate-surface bond when it comes to adsorption properties and also the energetics of corresponding chemical reactions.56 In this work, the hybrid DFT calculation (HSE06)57 was also performed to check the consistency of the result calculated using DFT-D2+Ud, in particular for C2H4 adsorbing on the Ovac site. Finally, the climbing image nudged elastic band method (CI-NEB) was utilized with four to eight transition images to determine the activation energy barriers for the initial C–H and CC bond cleaving of adsorbed C2H4. Vibrational frequency calculations were then used for the confirmation of the transition state (TS).
3 Results and discussion
3.1 Oxygen vacancy (Ovac) on anatase TiO2 (001) surface
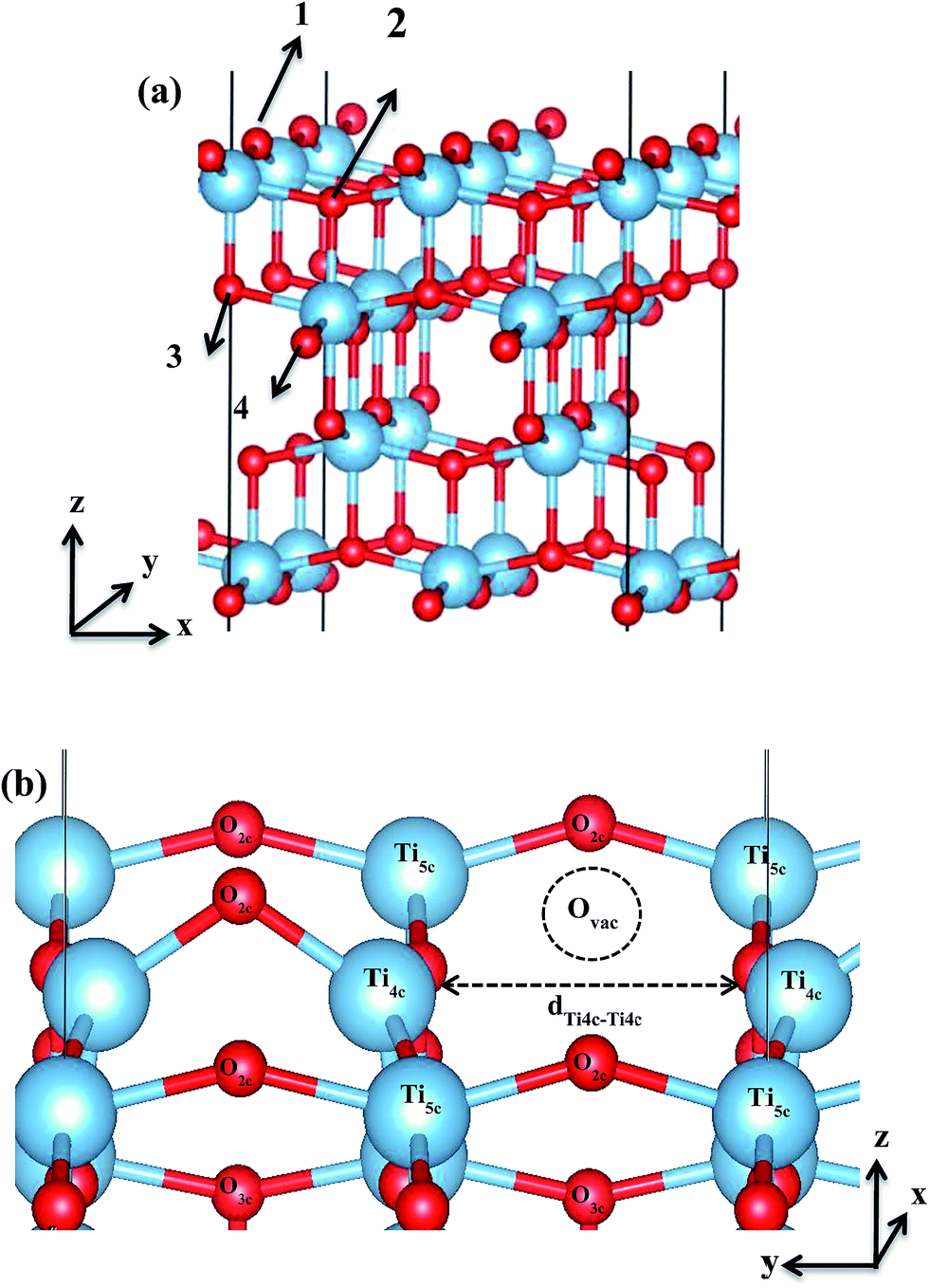
To determine the Ovac site on the unreconstructed anatase TiO2 (001), four different sites were considered, two on the surface and two in the sub-surface (Fig. 1). Here, ∼0.25 ML of vacancy concentration was utilized. The Ovac concentration considered gives 7.65 Å of Ovac–Ovac distance which leads to almost isolated impurities. The vacancy formation energies (Evac-formation) needed to remove the oxygen from each site were then calculated. Evac-formation is calculated by using the formula:| | |
Evac-formation = ETiO2−x − (ETiO2 + ½ EO2)
| (3) |
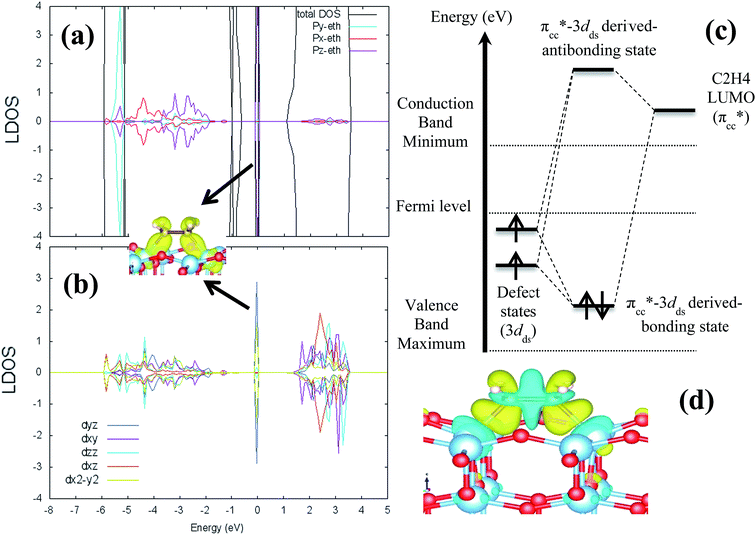
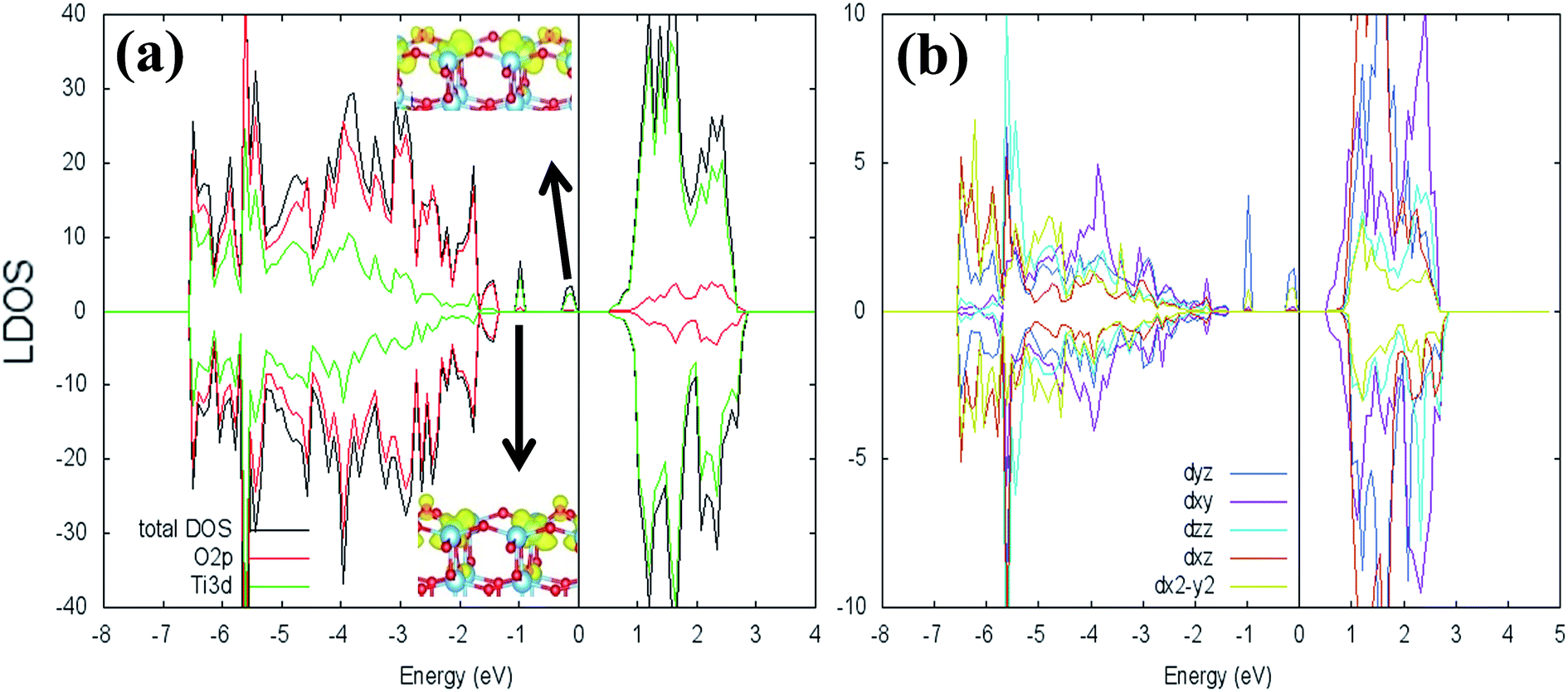
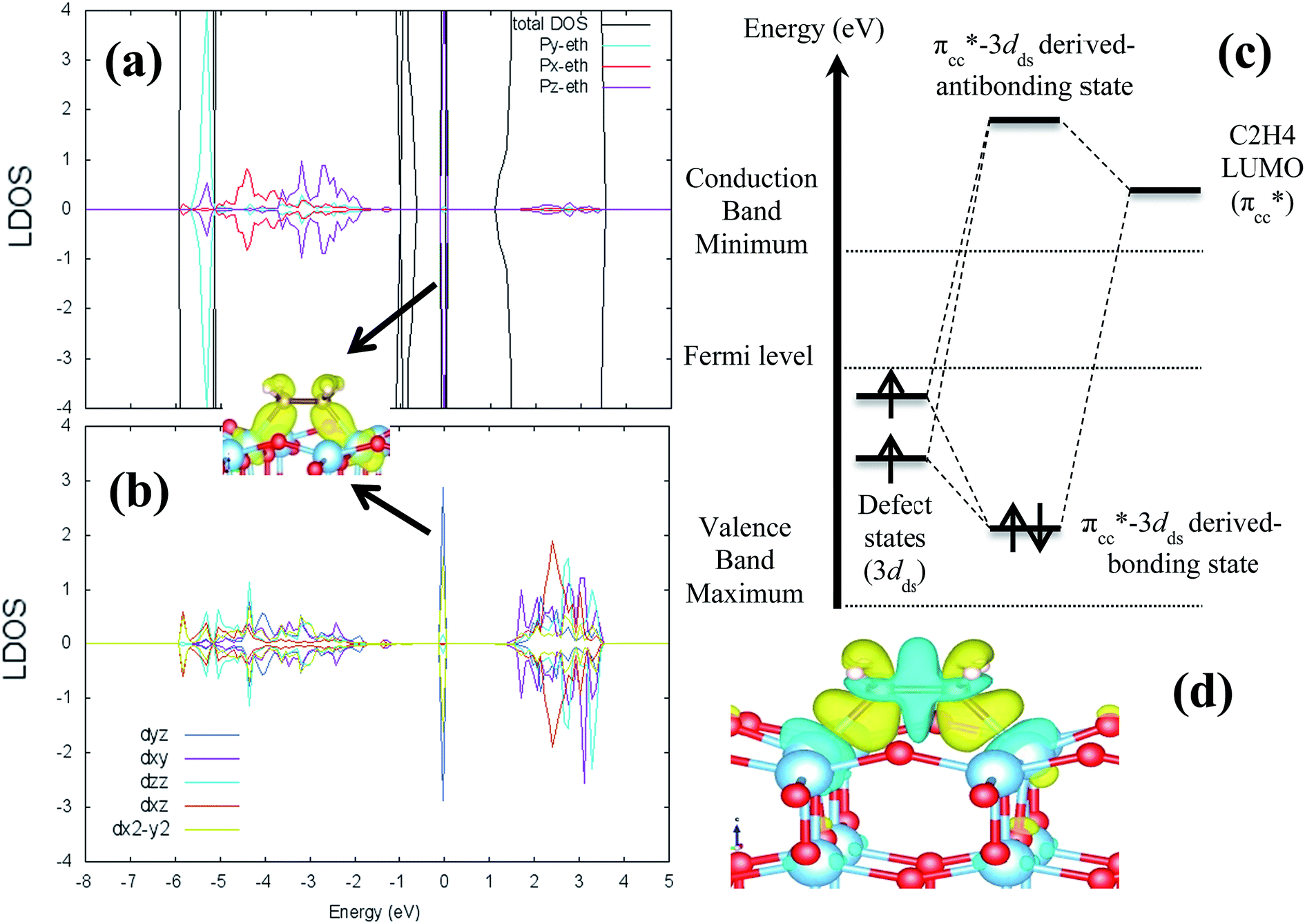
where ETiO2−x is the total energy of the reduced slab, ETiO2 is total energy of the pristine slab, and EO2 is the free energy of the oxygen molecule in the gas phase. Calculated vacancy formation energies (Table 1) show that two-coordinated oxygen (2-fold or O2c) on the surface is thermodynamically the most feasible site for Ovac formation (site #1). Upon the formation of surface Ovac, the two neighboring Ti atoms moved away from each other, resulting in an elongation of the Ti4c–Ti4c distance (dTi4c–Ti4c) to ∼4.89 Å (Ti–Ti atom distance on the pristine slab of (001) surface is ∼3.83 Å). The changes because of the oxygen vacancy also appeared in the electronic structure of the reduced surface. Local density of states (LDOS) of the defective surface shows the emergence of two defect states within the band-gap, located at around ∼0.5 eV and ∼1.2 eV from the conduction band minimum (Fig. 2a). These defect states can be attributed to the 3d conduction band of TiO2, being occupied by the excess electrons originating from the removed O. The charge density profile of the defect states shows accumulation of excess electrons on the two adjacent Ti atoms near the Ovac (Fig. 2b). This accumulation produces a polaronic type lattice distortion to the atoms located in the vicinity of the Ovac. Commonly, this particular distortion originates from the charge carriers which exist in the polar semiconductor or insulator, producing a repulsive interaction between ions with the same charge and attractive interactions between those with the opposite charge. In the present case, this interaction originated from the excess electrons of the Ovac that reside in the nearest Ti-atoms. This is similar to what has also been observed in a previous work58 [LDOS of TiO2 (001) with a surface Ovac for DFT-D2 and DFT-D2+Ud = 4 eV can be found in the ESI Fig. S1 and S2†]. Overall, the present result for the reduced anatase TiO2 (001) surface with Ovac shows good agreement with the results found for previous theoretical calculations58–60 as well as experimental observations using photoemission spectroscopy.61,62
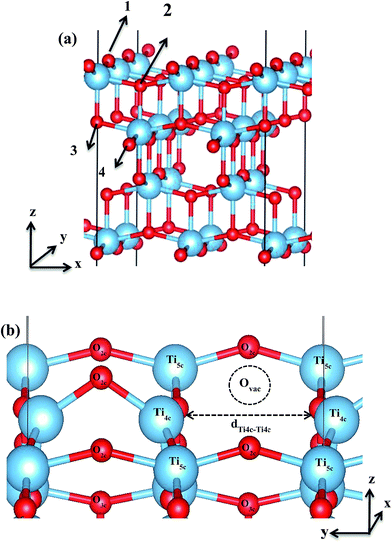 |
| | Fig. 1 (a) The numbers (1–4) indicate the four possible Ovac sites considered in this work, (b) Ovac site considered for C2H4 adsorption in this work and the surrounding area near the vacancy site [site #1 in (a)]. The dTi4c–Ti4c denotes the elongation of two 4-fold Ti (Ti4c) upon the C2H4 formation. | |
Table 1 Calculated neutral Ovac formation energy (Evac-formation)
| Site |
Evac-formation (eV) |
Difference |
| 1 |
3.50 |
— |
| 2 |
4.35 |
0.85 |
| 3 |
4.47 |
0.97 |
| 4 |
4.80 |
1.30 |
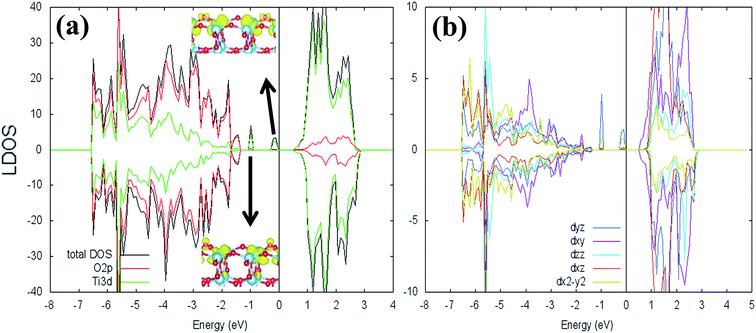 |
| | Fig. 2 Local density of states (LDOS) of (a) defective anatase TiO2 (001) with surface Ovac calculated DFT-D2+Ud (Ud = 3 eV), (b) 3d orbital projection of the two Ti atoms nearest to the vacancy site. Negative and positive values of the y-axis are minority and majority spin, respectively. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3). | |
3.2 C2H4 molecular adsorption on a surface with an Ovac
Previous studies have observed several geometric configurations for C2H4-solid surface interaction such as π-complex, di-σ complex, ethylidene, vinylidene, ethylidyne and ethylylidene.63 This study focused on the π-complex and di-σ complex interactions. These interactions have been consistently observed in the low temperature C2H4-solid surface experiments, in which C2H4 molecularly adsorbs parallel to the surface and remains intact with respect to its gas phase.64 The previously mentioned interactions mainly originate from the highly reactive π/π* orbitals of the C2H4 CC bond.
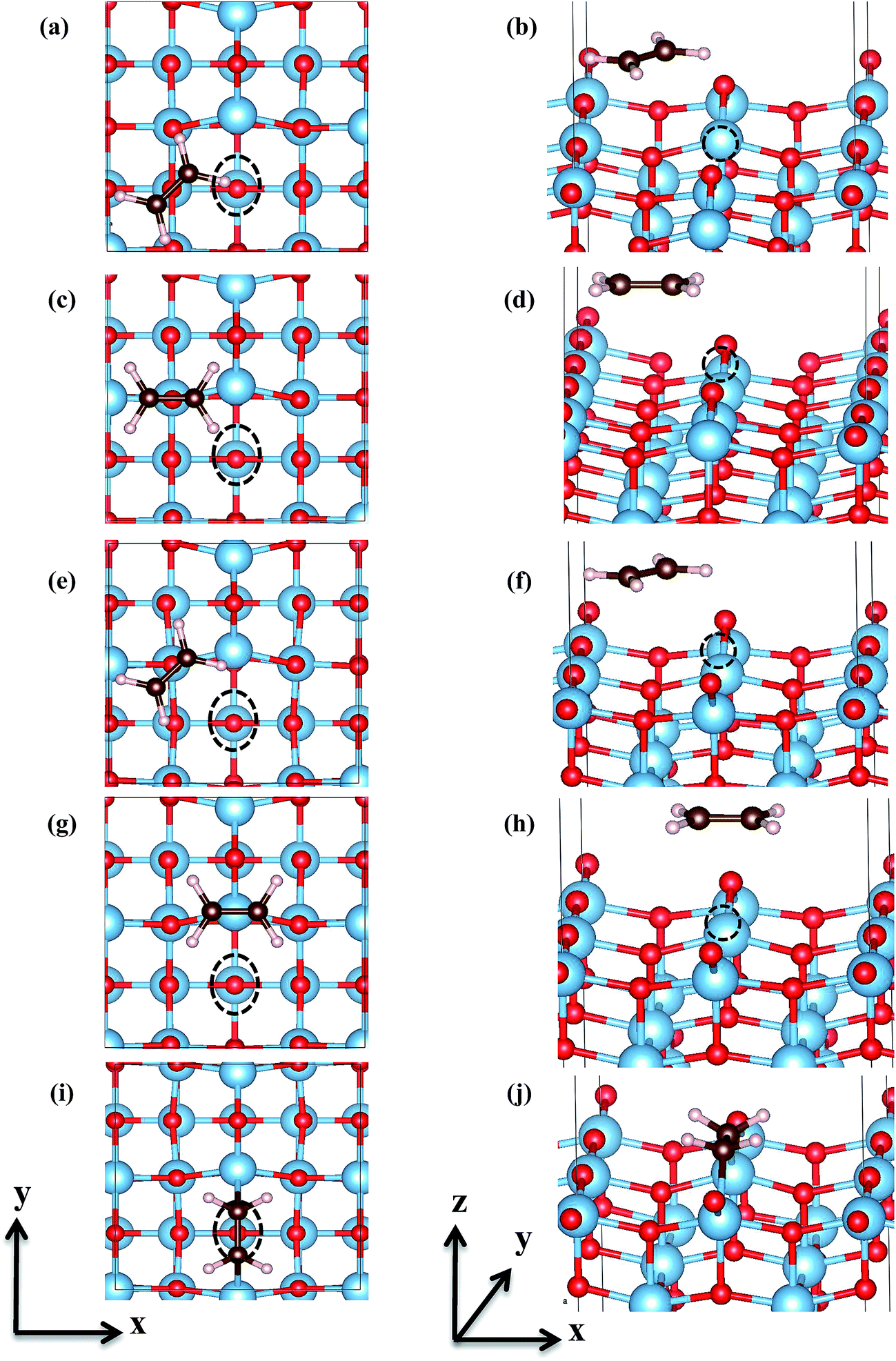
Five different adsorption configurations of C2H4 on the reduced anatase TiO2 (001) with surface Ovac as shown in Fig. 3 were studied. Note that only C2H4 adsorption on the reduced anatase TiO2 (001) with Ovac created at site #1 which is the most feasible site for creating Ovac was considered. Single point calculations were first performed for each of the five configurations, where all degrees of freedom were constrained and only the vertical distance of C2H4 relative to the surface was varied (Fig. S3, ESI†). Based on the one-dimensional potential energy curves obtained from each of the configurations, the distance at which the C2H4-reduced TiO2 (001) surface system gives the lowest energy was further fully optimized. In previous work by Shukri and Kasai, it was found that the long range dispersion interaction shows a significant contribution to the adsorption energy of C2H4. This affects overall C2H4 stability on the pristine TiO2 surface.15 The contribution of vdW interaction was also checked by comparing the calculated adsorption energy of C2H4 on each adsorption site. Indeed, the vdW interaction still contributes a significant part to the overall calculated adsorption energies (Table S1, ESI†). Therefore, in the present study, the long range dispersion interaction (vdW interaction) was also included in the calculations.
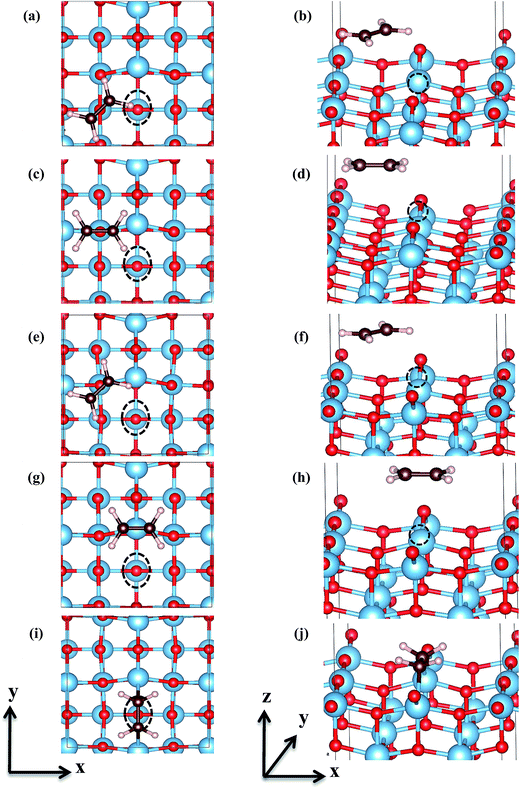 |
| | Fig. 3 Top view (left panels) and side view (right panels) of C2H4 adsorption configurations on and in the vicinity of surface Ovac, (a) and (b) are C2H4 on hollow-site [Hol], (c) and (d) are C2H4 on O3c-site [O3c], (e) and (f) are tilted-C2H4 on O3c-site [O3c-tilted], (g) and (h) are C2H4 on Ti4c-site [Ti4c], (i) and (j) are C2H4 on Ovac-site [Ovac]. The name for each configuration is given in square brackets. The dash–line circle corresponds to the location of oxygen vacancy. | |
The energetics and geometric properties of C2H4 adsorbed on a defective anatase TiO2 (001) surface calculated at DFT-D2+Ud (Ud = 3 eV) are shown in Table 2. C2H4 adsorption on Ti5c, O3c, hollow, and O3c-tilted sites resulted in relatively weak adsorption energies. These weak interactions were also indicated by the geometrical structure of C2H4, having almost no perturbation on its initial sp2-hybridization and adsorbed at a relatively far distance from the reduced TiO2 (001) surface. For molecularly adsorbed C2H4 on those four previously mentioned sites, it can be inferred that vdW is the dominant interaction that stabilizes the adsorption.
Table 2 Adsorption energies and selected geometric parameters of adsorbed C2H4 on different configurations calculated at DFT-D2+Ud (Ud = 3 eV). See Fig. 3 for the names of each adsorption configuration
| Configuration |
ΔEads (eV) |
dC–C (Å) |
dC–H (Å) |
dC–Ti (Å) |
| Ti4c |
−0.10 |
1.33 |
1.10 |
3.18 |
| O3c |
−0.18 |
1.33 |
1.09 |
3.32 |
| Ovac |
−0.94 |
1.47 |
1.09 |
2.10 |
| Hol |
−0.32 |
1.33 |
1.09 |
3.87 |
| O3c-tilted |
−0.15 |
1.33 |
1.10 |
3.40 |
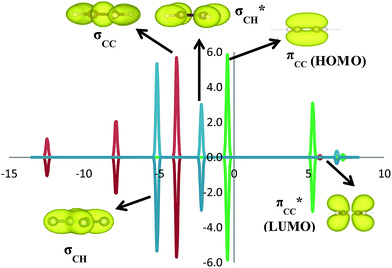
C2H4 adsorption on an O-vacancy site (O2c-site), however, has a quite distinct property when compared to the other four configurations. Upon adsorption, the initial (gas phase) sp2-hybridization of C2H4 changed into sp3-type structure, following the elongation of the C2H4 CC bond. The CC bond was elongated to ∼0.16 Å compared to its gas phase bond length, indicating a weakened bonding interaction between the two carbon atoms. This weakening also corresponds to the decrement of the C2H4 bond order from the initial double bond (CC) to become a single bond (C–C). From now on, the weakened carbon–carbon bond of adsorbed C2H4 with the C–C notation will be referred to. In order to further elucidate the mechanism of C2H4 adsorption on the Ovac-site, the orbital-resolved LDOS of the isolated C2H4 gas phase and C2H4 adsorbed on the Ovac-site [Fig. 4 and 5] was plotted. Here, the pz-orbitals of the two C atoms formed the highest occupied molecular orbital (HOMO; πcc)–LUMO 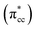 of C2H4. These two frontier molecular orbitals (MOs) often play a significant part in C2H4 interaction with various solid surfaces. By observing the plot of the orbital-resolved LDOS of C2H4 adsorbed on the Ovac site (Fig. 5), it can be seen that there is a strong hybridization between the p-states of C2H4 C–C with the Ti 3d-states, i.e., two adjacent Ti atoms in the vicinity of the Ovac.65 Another noticeable feature is the emergence of a localized occupied state below the Fermi level, which formed by the mixing of the
of C2H4. These two frontier molecular orbitals (MOs) often play a significant part in C2H4 interaction with various solid surfaces. By observing the plot of the orbital-resolved LDOS of C2H4 adsorbed on the Ovac site (Fig. 5), it can be seen that there is a strong hybridization between the p-states of C2H4 C–C with the Ti 3d-states, i.e., two adjacent Ti atoms in the vicinity of the Ovac.65 Another noticeable feature is the emergence of a localized occupied state below the Fermi level, which formed by the mixing of the 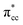 of C2H4 with the occupied Ti 3d defect states.66 This hybridization implies that charges from occupied defect states of Ti 3d are transferred to the
of C2H4 with the occupied Ti 3d defect states.66 This hybridization implies that charges from occupied defect states of Ti 3d are transferred to the 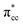 of C2H4, indicating a back donation from the defect states to C2H4 that subsequently weakened the adsorbed C2H4 C–C bond. The weakening because of the depletion of charges in the C–C region upon adsorption can also be observed, whereas accumulation of charges took place in the two C–Ti bonding and C–H bonding regions (Fig. 5d). The accumulation of charges in the C–H bonding regions subsequently tilted the four hydrogen atoms, and as a consequence the C2H4 minimized the repulsive interactions between its bonding electrons. Fig. 5 also shows the plot of the spin density profile within the energy range of Ti 3d defect states –
of C2H4, indicating a back donation from the defect states to C2H4 that subsequently weakened the adsorbed C2H4 C–C bond. The weakening because of the depletion of charges in the C–C region upon adsorption can also be observed, whereas accumulation of charges took place in the two C–Ti bonding and C–H bonding regions (Fig. 5d). The accumulation of charges in the C–H bonding regions subsequently tilted the four hydrogen atoms, and as a consequence the C2H4 minimized the repulsive interactions between its bonding electrons. Fig. 5 also shows the plot of the spin density profile within the energy range of Ti 3d defect states – 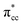 derived-bonding localized mid-gap state that shows two σ-type bonding profiles between both C atoms of C2H4 with the two Ti atoms near the vacancy site. These two σ-type interactions (or the so-called di-σ complex) characterize the rehybridization of C2H4 into the sp3-type upon its adsorption on the Ovac site. It is interesting to note that previous experimental work has shown that coating a anatase TiO2 surface using C2H4 and acetylene as carbon sources resulted in the decrease of the TiO2 band-gap by about ∼0.7 eV, subsequently increasing the photoactivity of the C-modified TiO2 under visible light.22 These present results clarified at least one possible origin of this band-gap decrement. The hybrid
derived-bonding localized mid-gap state that shows two σ-type bonding profiles between both C atoms of C2H4 with the two Ti atoms near the vacancy site. These two σ-type interactions (or the so-called di-σ complex) characterize the rehybridization of C2H4 into the sp3-type upon its adsorption on the Ovac site. It is interesting to note that previous experimental work has shown that coating a anatase TiO2 surface using C2H4 and acetylene as carbon sources resulted in the decrease of the TiO2 band-gap by about ∼0.7 eV, subsequently increasing the photoactivity of the C-modified TiO2 under visible light.22 These present results clarified at least one possible origin of this band-gap decrement. The hybrid 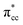 – 3d defect state resulting from C2H4 – reduced anatase TiO2 (001) coupling (located at ∼1.0 eV below the conduction band minimum) can account for the increasing photoactivity of C-modified TiO2, thus extending the system photoactivity to the visible light range. Thus, exposing the system to ultraviolet-visible light can trigger electronic excitation from the hybrid
– 3d defect state resulting from C2H4 – reduced anatase TiO2 (001) coupling (located at ∼1.0 eV below the conduction band minimum) can account for the increasing photoactivity of C-modified TiO2, thus extending the system photoactivity to the visible light range. Thus, exposing the system to ultraviolet-visible light can trigger electronic excitation from the hybrid 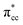 – 3d defect state to the conduction band that accounts for the experimentally observed decreasing band-gap. This is similar to other electron-acceptor molecules adsorbed on a reduced TiO2 surface such as oxygen (O2), where a localized mid-gap state was also observed upon adsorption.67 In addition, a hybrid-DFT calculation was also performed to check the consistency of the present result (C2H4 adsorbed on the Ovac-site) obtained by DFT+Ud (Ud = 3 eV). The optimized geometry and adsorption energy results obtained using hybrid-DFT are in good agreement with the results obtained from DFT+Ud (Ud = 3 eV) (Table 3). This consistency ensures the reliability of the chosen Ud value. To this end, it can be concluded that excess electrons because of the oxygen vacancy play a vital role to stabilize C2H4 adsorption on the reduced anatase TiO2 (001) surface. The present finding is in contrast to the C2H4 adsorption on clean/pristine anatase TiO2 (001), in which C2H4 adsorption is mainly driven by the dispersion (or vdW) interaction.15
– 3d defect state to the conduction band that accounts for the experimentally observed decreasing band-gap. This is similar to other electron-acceptor molecules adsorbed on a reduced TiO2 surface such as oxygen (O2), where a localized mid-gap state was also observed upon adsorption.67 In addition, a hybrid-DFT calculation was also performed to check the consistency of the present result (C2H4 adsorbed on the Ovac-site) obtained by DFT+Ud (Ud = 3 eV). The optimized geometry and adsorption energy results obtained using hybrid-DFT are in good agreement with the results obtained from DFT+Ud (Ud = 3 eV) (Table 3). This consistency ensures the reliability of the chosen Ud value. To this end, it can be concluded that excess electrons because of the oxygen vacancy play a vital role to stabilize C2H4 adsorption on the reduced anatase TiO2 (001) surface. The present finding is in contrast to the C2H4 adsorption on clean/pristine anatase TiO2 (001), in which C2H4 adsorption is mainly driven by the dispersion (or vdW) interaction.15
 |
| | Fig. 4 Isolated molecular orbital (MO) of C2H4 projected onto the sum of the two C atoms' 2p-state with their corresponding orbitals. The x-axis represents energy in eV and the y-axis corresponds to the states per energy level (isosurface = 0.02 e Å−3). | |
 |
| | Fig. 5 LDOS of C2H4 adsorbed on a Ovac-site calculated at DFT-D2+Ud (Ud = 3 eV) and the corresponding partial charge density of the newly formed mid-gap state. (a) Orbital-resolved LDOS of the two C-atoms of C2H4 projected onto their p-state, plotted together with total DOS, (b) orbital-resolved LDOS of the two Ti-atom near the Ovac projected onto their 3d-orbital, (c) simplified schematic interaction between 3d defect states (3ddef) with C2H4 LUMO 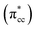 , (d) charge density difference (CDD) plot of C2H4 adsorbed on Ovac-site. The green color corresponds to the charge depleted region, whereas the yellow corresponds to the charge accumulated region. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3). , (d) charge density difference (CDD) plot of C2H4 adsorbed on Ovac-site. The green color corresponds to the charge depleted region, whereas the yellow corresponds to the charge accumulated region. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3). | |
Table 3 Calculated adsorption energies and selected geometrical parameters of C2H4 adsorbed on the Ovac-site calculated at DFT-D2+Ud (Ud = 4 eV, 5 eV) and hybrid-DFT
| Calculation method |
Eads (eV) |
C–C (Å) |
C–H (Å) |
C–Ti (Å) |
| DFT-D2+Ud (Ud = 4 eV) |
−0.8 |
1.49 |
1.1 |
2.14 |
| DFT-D2+Ud (Ud = 5 eV) |
−0.32 |
1.35 |
1.1 |
2.64 |
| Hybrid-DFT (HSE06) |
−1.12 |
1.50 |
1.10 |
2.10 |
Previous research has shown that by controlling the work function (W), it may be possible to control the redox chemistry of oxide materials as well.25,28 In conjunction with the present work on the C2H4-reduced TiO2 system, the influence of increasing reduced anatase TiO2 (001) work function (WTiO2) on the adsorption of C2H4 was also checked. This idea is tested in an ad hoc manner, whereby the position of the defect state (which simultaneously determines the WTiO2) is varied upon altering the Ud values. Here, the C2H4 adsorption properties on Ovac for two other Ud values (Ud = 4 eV and 5 eV) have been calculated. The different behaviors of C2H4 adsorption calculated with Ud = 5 eV were observed, whereas the result for Ud = 4 eV showed overall similarity to the result calculated with Ud = 3 eV (Fig. S4, ESI†). C2H4 retained its planar geometry and its adsorption energy was also significantly lowered (Table 3). In order to further elucidate the decrease of the adsorption strength of C2H4 on the vacancy site calculated with Ud = 5 eV, the LDOS for the present case were plotted (Fig. 6a). Compared to the DFT-D2+Ud (Ud = 3 eV and 4 eV) cases, some less significant orbital hybridization can be observed, in particular between the Ti 3d defect states with C2H4 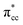 (LUMO). Indeed, some fractions of C2H4
(LUMO). Indeed, some fractions of C2H4 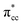 can still be observed within the gap states and also near the valence band edge, showing some electronic filling of the
can still be observed within the gap states and also near the valence band edge, showing some electronic filling of the 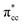 orbital. However, those hybridizations are not strong enough to produce significant stabilization for the C2H4-reduced TiO2 (001) coupling, because most of the broadening of
orbital. However, those hybridizations are not strong enough to produce significant stabilization for the C2H4-reduced TiO2 (001) coupling, because most of the broadening of 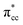 still occupies the region above the Fermi level, resulting in less significant interplay with the πcc (HOMO). This indicates that charge transfer from the surface to the C2H4 is significantly hindered because of the deeper emergence of the defect states in the energy level (or more tightly bound excess electrons) (Fig. 6b). Nevertheless, the tendency of the di-σ bonding formation from the charge density difference (CDD) plot can still be seen, as the charges accumulate in both the C–Ti regions (Fig. 6c). The results obtained from DFT-D2+Ud (Ud = 5 eV) imply another important point that the deeper the energy level of excess electrons (or the more the excess electrons are tightly bound), the more difficult the charge transfer to the C2H4 LUMO will be. This is similar to if the work function of a substrate is controlled, as has also been demonstrated by previous work of O2 adsorption on reduced rutile TiO2 (110) surface.29
still occupies the region above the Fermi level, resulting in less significant interplay with the πcc (HOMO). This indicates that charge transfer from the surface to the C2H4 is significantly hindered because of the deeper emergence of the defect states in the energy level (or more tightly bound excess electrons) (Fig. 6b). Nevertheless, the tendency of the di-σ bonding formation from the charge density difference (CDD) plot can still be seen, as the charges accumulate in both the C–Ti regions (Fig. 6c). The results obtained from DFT-D2+Ud (Ud = 5 eV) imply another important point that the deeper the energy level of excess electrons (or the more the excess electrons are tightly bound), the more difficult the charge transfer to the C2H4 LUMO will be. This is similar to if the work function of a substrate is controlled, as has also been demonstrated by previous work of O2 adsorption on reduced rutile TiO2 (110) surface.29
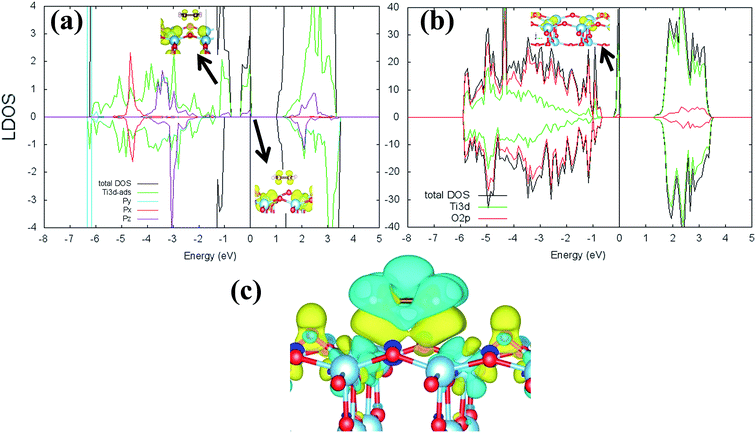 |
| | Fig. 6 (a) Orbital-resolved LDOS of two C-atoms of C2H4 projected onto their 2p-state plotted together with total DOS calculated at DFT-D2+Ud (Ud = 5 eV). Projection of partial charge density profile of 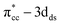 within two energy windows are also shown, (b) LDOS of reduced anatase TiO2 (001) with Ovac calculated at DFT-D2+(Ud = 5 eV), and (c) CDD plot of C2H4 adsorbed on Ovac-site. Green color corresponds to charge depleted region, whereas yellow corresponds to charge accumulated region. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3). within two energy windows are also shown, (b) LDOS of reduced anatase TiO2 (001) with Ovac calculated at DFT-D2+(Ud = 5 eV), and (c) CDD plot of C2H4 adsorbed on Ovac-site. Green color corresponds to charge depleted region, whereas yellow corresponds to charge accumulated region. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3). | |
3.3 C2H4 dissociative adsorption on a surface with an O-vacancy
In this sub-section, the relevant results for two initial decomposition pathways of C2H4 will be presented, namely: (1) C–H bond cleaving (or dehydrogenation), and (2) direct C–C bond cleaving.| |
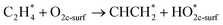 | (4) |
| |
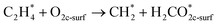 | (5) |
The ‘*’-symbol indicates the adsorbed species on the anatase TiO2 (001) surface; O2c-surf corresponds to the nearest two-fold coordinated oxygen (from the Ovac-site) on the topmost layer. Note that adsorbed C2H4 on Ovac-site was adopted as the initial configuration for both decomposition pathways. The products for the two initial decomposition pathways are determined based on the two criteria: (1) the adsorbed C2H4 dissociated fragments that give the most energetically stable configuration (relative to isolated gas-phase C2H4 and reduced TiO2 (001) slab); (2) the shortest diffusion pathway for C2H4 H-atom upon dehydrogenation. The first pathway (dehydrogenation) results in the breaking of one H atom of C2H4, followed by the formation of a surface hydroxyl with the nearest O2c (two-fold coordination oxygen), whereas the remaining fraction (CHCH2 or vinyl) makes a bond with the two Ti atoms nearest to the Ovac-site. For the second pathway (C–C bond cleaving), C2H4 breaks into two CH2 fragments, where one fragment occupies the Ovac-site and another fragment forms adsorbed formaldehyde with the nearest neighbor O2c (two-fold coordination oxygen) as well. In addition, the consideration of formaldehyde as a C–C cleaving product is based on the previous experimental work in which formaldehyde is frequently observed during hydrocarbon–oxide interaction.68,69
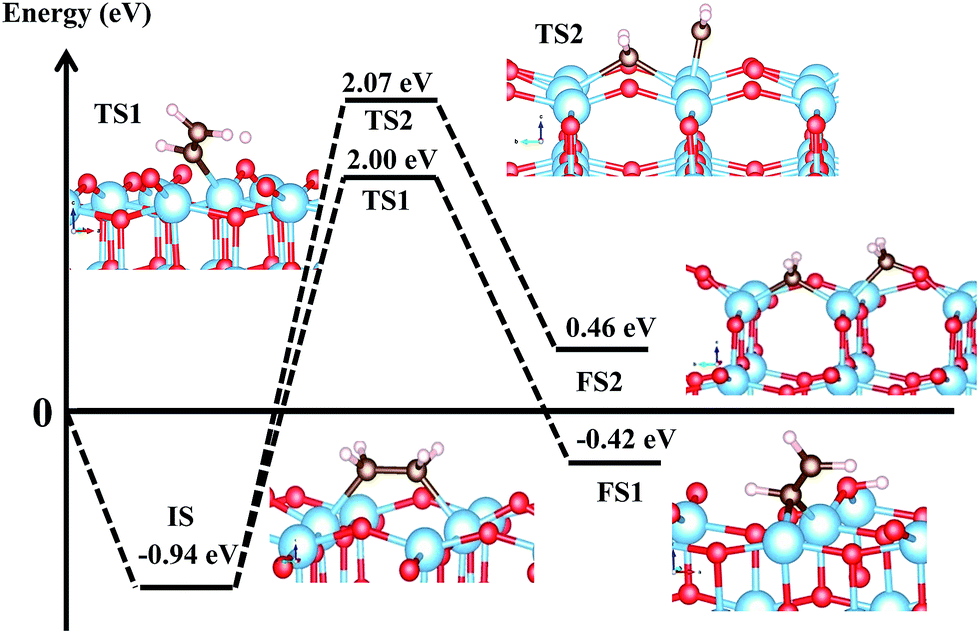
As shown in Fig. 7, it can be see that both decomposition pathways are highly activated reactions with calculated activation barriers of 2.94 eV and 3.01 eV for C–H and C–C bond cleaving, respectively. The calculated reaction energies for both decomposition pathways show similar endothermic characteristics. It is interesting to see that both decomposition pathways show almost similar activation energies. We can attribute this similarity in high activation barrier, partly, to the fundamental characteristics of the C–H and C–C bonds. For dehydrogenation, note that the C–H bond is formed by a strong σ-type interaction, so that breaking this particular bond requires a large amount of energy. It is also important to note that diffusion energy needed by the H to move to the nearest O2c (to form the surface hydroxyl) is also accounted for in the present calculated barrier. This is also in agreement with results from previous work, where atomic H needs to overcome a certain amount of activation barriers to diffuse on the surface of TiO2, and this can range from 0.7 eV–2.06 eV, depending on the diffusion pathways and simulation environments.70,71
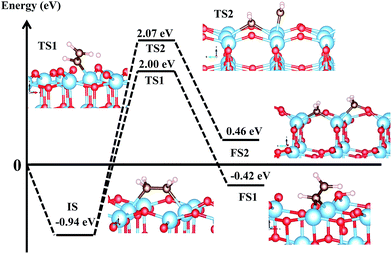 |
| | Fig. 7 First two initial decomposition pathways of C2H4 adsorbed on Ovac-site. Reaction (4) corresponds to C2H4 dehydrogenation, whereas (5) corresponds to direct CC bond cleaving. IS, TS, and FS correspond to initial, transition, and final state, respectively. All energies are calculated with respect to isolated (gas phase) C2H4 and reduced slab anatase TiO2 (001) with surface Ovac. | |
For C–C bond cleaving, a slightly higher activation barrier by about 0.07 eV can be observed, when compared to the C–H bond cleaving. However, similar to the C–H bond, the same argument also prevails in the C–C bond cleaving case, because, once again, the breaking of the σ-type interaction is being dealt with. The C–C bond cleaving consequently requires a certain amount of energy as well. Another important point is that the activation barrier for the C–C bond cleaving also involves the contribution from the energy needed to break the Ti4c–O2c bonding nearest to the Ovac-site, in order to facilitate the formation of adsorbed formaldehyde species. To this end, it can be inferred that this high activation barrier for C–C bond cleaving stems from two contributions: the energy needed to break the C–C σ-bond, and also, subsequently, the Ti4c–O2c bond in order to facilitate H2CO formation as the final product. To some extent, this similarity is analogous to C–H and C–C bond breaking of gas phase C2H6 in which all the CC and CH bonds are composed by σ-type bonding (the energies needed to break CC and CH in C2H6 are ∼3.95 eV and ∼4.36 eV, respectively). Indeed, to the best of our knowledge, there are direct experimental data available that can be referred to and compared with the present results. However, the present tendency of the formation of C–C coupling on the Ovac is similarly observed when reduced rutile TiO2 (110) with surface bridging Ovac is exposed to formaldehyde. By using scanning tunneling microscopy, Zhu et al. observed that, mediated by the Ovac, adsorbed formaldehyde and/or methylene can couple with other adsorbed formaldehyde/methylene species on the surface through several reaction channels, forming C2H4 as the final product as the temperature increased.68,69 The present results support these experimental findings, in which coupled C–C bond (or described here as molecularly adsorbed C2H4) is more favorable when compared to the dissociative adsorption cases when Ovac exists on the surface of TiO2 [as shown by the high dissociation barriers and the comparison of thermodynamic stability of the initial reactant (molecularly adsorbed C2H4) with the final product, namely, C–H and C–C bond cleaving]. An interesting recent work on acetylene (C2H2) adsorption on anatase TiO2 (101) and (001) surfaces by Chen et al. should also be mentioned briefly.72 The published report focuses on the study of C2H2 cyclo-oligomerisation mechanism on anatase TiO2 surfaces. In particular, it was reported that C2H2 spontaneously dissociates upon adsorption on the pristine anatase TiO2 (001) via H–CCH bond cleaving. The dissociated CCH fragment then binds to the surface of the Ti, forming a Ti–CCH surface complex where the H binds to the nearest O2c producing surface OH. Therefore, there seems to be a contradiction between the present results with those obtained by Chen et al., as a relatively high activation energy of C2H4 CH bond cleaving was reported in the current paper. However, it is also important to note some aspects of the two systems in order to come to terms with these seemingly different results. The present results deal with reduced anatase TiO2 (001) with surface Ovac whereas Chen et al. deal with the pristine surface of anatase TiO2 (001). This difference in the surface of interest may already lead to a different adsorption and reaction mechanism as well. A qualitative (and to some extent semi-quantitative) similarity that may be noticed is that in terms of the thermodynamics of the reaction, this result agrees with that obtained by Chen et al., in which reduced anatase TiO2 (001) favors the dehydrogenation or the CH bond cleaving. Indeed, it is more reasonable and interesting to further discuss the C2H2 adsorption on the pristine anatase TiO2 (001) results with the previous work of C2H4 on pristine anatase TiO2 (001).15 However, this is already beyond the scope of the present work. More rigorous work related to the decomposition pathways of unsaturated hydrocarbon (both C2H2 and C2H4) on the pristine (as well as hydroxylated) surface of anatase surfaces is currently being done and will soon be covered in a future report.
4 Conclusion
Based on density functional theory calculations, C2H4 adsorption on an anatase TiO2 (001) surface with Ovac has been studied. Oxygen vacancies tend to form on the topmost layer, occupying the O2c (2-coordinated oxygen ion) site with a vacancy formation energy of ∼3.5 eV. The results presented in this paper suggest that C2H4 adsorbed on the Ovac-site is the most stable configuration. The adsorption of C2H4 on the Ovac-site is accompanied by C2H4 rehybridization from its initial gas-phase sp2 structure to sp3-type hybridization. This is because of the filling of the C2H4 LUMO by the excess electrons of the Ovac residing in the vicinity of the two nearest adjacent Ti atoms. This implies that the presence of an Ovac is important for C2H4 binding on the TiO2 surface. Electronic structure analysis shows that the bonding between C2H4 and surface Ti atoms on the Ovac site produced a new localized mid-gap state. This state could provide a direct explanation for the origin of the reduced band-gap observed in a previous optical measurement of carbon-coated TiO2. Subsequent calculations on two initial decomposition pathways of C2H4 adsorbed on the Ovac-site show that the C–H bond and C–C bond cleaving require high activation barriers, with the C–H bond cleaving being slightly more favorable when compared to the CC bond cleaving (2.94 eV and 3.01 eV, respectively) and the final states of the two initial decomposition pathways show similar endothermic characteristics. Finally, this finding indicates that surface Ovac-site tends to promote the formation of molecularly adsorbed Ti-bound sp3-C2H4 (–Ti–CH2CH2–Ti–) when compared to the dissociative adsorption case.
Acknowledgements
This work is supported in part by: Japan Ministry of Education, Culture, Sports, Science, and Technology (MEXT) Grant-in-Aid for Scientific Research (15H05736, 24246013, 15KT0062, 26248006); NEDO Project “R&D Towards Realizing an Innovative Energy Saving Hydrogen Society based on Quantum Dynamics Applications”; and the Osaka University Joining and Welding Research Institute Cooperative. Some of the numerical calculations presented here were done using the computer facilities at the following institutes: CMC (Osaka University), ISSP, KEK, NIFS, and YITP. GS would like to express gratitude to MEXT for the financial study aid. HKD and MKA gratefully acknowledge research grant support from the Institut Teknologi Bandung – Japan International Cooperation Agency (ITB-JICA) under the “Proyek Pengembangan ITB (III)” scheme. Fruitful discussions with Dr Adhitya Gandaryus Saputro (Institute of Technology, Bandung) and Dr Allan A. B. Padama (University of the Philippines, Los Baños) are also acknowledged.
References
- M. Baldi, V. S. Escribano, J. M. G. Amores, F. Milella and G. Busca, Appl. Catal., B, 1998, 17, L175 CrossRef CAS.
- M. Paulis, L. M. Gandia, A. Gil, J. Sambeth, J. A. Odriozola and M. Montes, Appl. Catal., B, 2000, 26, 37 CrossRef CAS.
- L. Forni and I. Rosetti, Appl. Catal., B, 2002, 38, 29 CrossRef CAS.
- M. R. Morales, B. P. Barbero and L. E. Cadús, Appl. Catal., B, 2007, 74, 1 CrossRef CAS.
- J. C. Mol, Catal. Today, 1999, 51, 289 CrossRef CAS.
- K. Tanaka, K. Miyahara and K. I. Tanaka, J. Mol. Catal., 1982, 15, 133 CrossRef CAS.
- M. Hussain, S. Bensaid, F. Geobaldo, G. Saracco and N. Russo, Ind. Eng. Chem. Res., 2011, 50, 2536 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788 CrossRef PubMed.
- J. Liu, Y. T. Kuo, K. J. Klabunde, C. Rochford, J. Wu and J. Li, ACS Appl. Mater. Interfaces, 2009, 1, 1645 CAS.
- G. K. Mor, S. Kim, M. Paulose, O. K. Varghese, K. Shankar, J. Basham and C. A. Grimes, Nano Lett., 2009, 9, 4250 CrossRef CAS PubMed.
- I. S. McLintock and M. Ritchie, Trans. Faraday Soc., 1965, 61, 1007 RSC.
- G. Busca, G. Porcile and V. Lorenzelli, J. Mol. Struct., 1986, 141, 395 CrossRef CAS.
- G. Busca, V. Lorenzelli, G. Ramis and V. Sanchez Escribano, Mater. Chem. Phys., 1991, 29, 175 CrossRef CAS.
- G. Busca, V. Lorenzelli, G. Ramis, J. Saussey and J. C. Lavalley, J. Mol. Struct., 1992, 267, 315 CrossRef CAS.
- G. Shukri and H. Kasai, Surf. Sci., 2014, 619, 59 CrossRef CAS.
- X. Fu, L. A. Clark, W. A. Zeltner and M. A. Anderson, J. Photochem. Photobiol., A, 1996, 97, 181 CrossRef CAS.
- S. Yamazaki, S. Tanaka and H. Tsukamoto, J. Photochem. Photobiol., A, 1999, 121, 55 CrossRef CAS.
- D.-R. Park, J. Zhang, K. Ikeue, H. Yamashita and M. Anpo, J. Catal., 1999, 185, 114 CrossRef CAS.
- T. A. Westrich, K. A. Dahlberg, M. Kaviany and J. W. Schwank, J. Phys. Chem. C, 2011, 115, 16537 CAS.
- B. Hauchecorne, T. Tytgat, S. W. Verbruggen, D. Hauchecorne, D. Terrens, M. Smits, K. Vinken and S. Lenaerts, Appl. Catal., B, 2011, 105, 111 CrossRef CAS.
- N. K. Memon, D. H. Anjum and S. H. Chung, Combust. Flame, 2013, 160, 1848 CrossRef CAS.
- D. H. Anjum, N. K. Memon and S. H. Chung, Mater. Lett., 2013, 108, 134 CrossRef CAS.
- S. M. Jain, J. J. Biedrzycki, V. Maurino, A. Zecchina, L. Mino and G. Spoto, J. Mater. Chem. A, 2014, 2, 12247 CAS.
- J. J. Biedrzycki, S. Livraghi, I. Corazzari, L. Mino, G. Spoto and E. Giamello, Langmuir, 2015, 31, 569 CrossRef CAS PubMed.
- M. T. Greiner, M. G. Helander, W. M. Tang, Z. B. Wang, J. Qiu and Z. H. Lu, Nat. Mater., 2012, 11, 76 CrossRef CAS PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185 CrossRef CAS.
- I. Justicia, P. Ordejón, G. Canto, J. L. Mozos, J. Fraxedas, G. A. Battiston, R. Gerbasi and A. Figueras, Adv. Mater., 2002, 14, 1399 CrossRef CAS.
- M. T. Greiner and Z. H. Lu, NPG Asia Mater., 2013, 5, e55 CrossRef CAS.
- N. A. Deskins, R. Rousseau and M. Dupuis, J. Phys. Chem. C, 2010, 114, 5891 CAS.
- A. S. Barnard and P. Zapol, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235403 CrossRef.
- A. S. Barnard, P. Zapol and L. A. Curtiss, Surf. Sci., 2005, 582, 173 CrossRef CAS.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. Lu and H. M. Cheng, J. Phys. Chem. C, 2009, 113, 21784 CAS.
- D. N. Pei, L. Gong, A. Y. Zhong, X. Zhang, J. J. Chen, Y. Mu and H. Q. Yu, Nat. Commun., 2015, 6, 8696 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638 CrossRef CAS PubMed.
- S. Lu, J. Yu and M. Jaroniec, Chem. Mater., 2011, 23, 4085 CrossRef.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078 CrossRef CAS PubMed.
- G. Kreese and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef.
- G. Kreese and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef.
- G. Kreese and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. Fahmi, C. Minot, B. Silvi and M. Causa, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 11717 CrossRef CAS.
- C. J. Horward, T. M. Sabine and F. Dickson, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 462468 Search PubMed.
- A. Vittadini and M. Casarin, Theor. Chem. Acc., 2008, 120, 551 CrossRef CAS.
- X. Q. Gong and A. Selloni, J. Phys. Chem. B, 2005, 109, 19560 CrossRef CAS PubMed.
- A. Hussain, J. Gracia, B. E. Nieuwenhuys and J. W. Niemantsverdriet, ChemPhysChem, 2010, 11, 2375 CrossRef CAS PubMed.
- V. M. Bermudez, J. Phys. Chem. C, 2011, 115, 6741 CAS.
- S. Grimme, J. Comput. Chem., 2006, 15, 1787 CrossRef PubMed.
- J. R. B. Gomez, J. P. Prates Ramalho and F. Illas, Surf. Sci., 2010, 604, 428 CrossRef.
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Phys. Rev. Lett., 2008, 100, 146401 CrossRef PubMed.
- B. J. Morgan and G. W. Watson, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 233102 CrossRef.
- E. Finazzi, C. D. Valentin, G. Pacchioni and A. Selloni, J. Chem. Phys., 2008, 129, 154113 CrossRef PubMed.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767 CrossRef CAS.
- B. Himmetoglu, A. Floris, S. Gironcoli and M. Cococcioni, Int. J. Quantum Chem., 2014, 114, 14 CrossRef CAS.
- M. García-Mota, A. Vojvodic, F. Abild-Pedersen and J. K. Nørskov, J. Phys. Chem. C, 2013, 117, 460 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- Y. Ortega, D. F. Hevia, J. Oviedo and M. A. San-Miguel, Appl. Surf. Sci., 2014, 294, 42 CrossRef CAS.
- T. Yamamoto and T. Ohno, Phys. Chem. Chem. Phys., 2012, 14, 589 RSC.
- B. J. Morgan and G. W. Watson, J. Phys. Chem. C, 2010, 114, 2321 CAS.
- A. G. Thomas, W. R. Flavell, A. R. Kumarasinghe, A. K. Mallick, D. Tsoutsou, G. C. Smith, R. Stockbauer, S. Patel, M. Grätzel and R. Hengerer, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035110 CrossRef.
- A. G. Thomas, W. R. Flavell, A. K. Mallick, A. R. Kumarasinghe, D. Tsoutsou, N. Khan, C. Chatwin, S. Rayner, G. C. Smith, R. L. Stockbauer, S. Warren, T. K. Johal, S. Patel, D. Holland, A. Taleb and F. Wiame, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035105 CrossRef.
- C. De La Cruz and N. Sheppard, J. Chem. Soc., Faraday Trans., 1997, 93, 3569 RSC.
- A. Kokalj, A. Dal Corso, S. Gironcoli and S. Baroni, J. Phys. Chem. B, 2006, 110, 367 CrossRef CAS PubMed.
- By further resolving the p-states of CC molecular orbital into its components (Fig. 5a), we can observe that it is not only the HOMO–LUMO of the ethylene (pz-state) that hybridize with Ti 3d-states, but also the px and py. Hence resulting in an overall strong resonance between the p-states of CC bond of ethylene with the adjacent two Ti 3d-state atoms near Ovac.
- LDOS projected onto each orbital components of both Ti 3d and ethylene CC molecular orbital shows this localized state is formed by the mixing of
 derived-bonding of C2H4 with the dyz, dx2–y2, and dzz orbitals of the Ti 3d defect states (Fig. 5a and b). It also can be seen that dyz orbital is the most dominant interacting state as compared to dx2–y2, and dzz. Two factors are the origin: (i) excess electrons from O-vacancy mostly occupy dyz-orbital, (ii) symmetry-wise, dyz-orbital has good compatibility with –in particular– pz-anti bonding
derived-bonding of C2H4 with the dyz, dx2–y2, and dzz orbitals of the Ti 3d defect states (Fig. 5a and b). It also can be seen that dyz orbital is the most dominant interacting state as compared to dx2–y2, and dzz. Two factors are the origin: (i) excess electrons from O-vacancy mostly occupy dyz-orbital, (ii) symmetry-wise, dyz-orbital has good compatibility with –in particular– pz-anti bonding 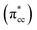 orbital of ethylene's CC.
orbital of ethylene's CC. - N. H. Linh, N. T. Quang, W. A. Diño and H. Kasai, Surf. Sci., 2015, 633, 38 CrossRef.
- K. Zhu, Y. Xia, M. Tang, Z. Wang, I. Lyubinetsky, Q. Ge, Z. Dohnálek, K. T. Park and Z. Zhang, J. Phys. Chem. C, 2015, 119, 18452 CAS.
- K. Zhu, Y. Xia, M. Tang, Z. Wang, B. Jan, I. Lyubinetsky, Q. Ge, Z. Dohnálek, K. T. Park and Z. Zhang, J. Phys. Chem. C, 2015, 119, 14267 CAS.
- M. M. Islam, M. Calatayud and G. Pacchioni, J. Phys. Chem. C, 2011, 115, 6809 CAS.
- U. Aschauer and A. Selloni, Phys. Chem. Chem. Phys., 2012, 14, 16595 RSC.
- H.-Y. T. Chen, S. Livraghi, E. Giamello and G. Pacchioni, ChemPlusChem, 2016, 81, 64 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra13633h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
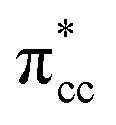 of C2H4, and simultaneously enhance the binding of C2H4 to the reduced anatase TiO2 (001). The bonding between the two C2H4 C atoms and the two Ti atoms nearest to the Ovac-site produces two σ-type bonds, leading to the emergence of a new localized mid-gap state. This hybrid
of C2H4, and simultaneously enhance the binding of C2H4 to the reduced anatase TiO2 (001). The bonding between the two C2H4 C atoms and the two Ti atoms nearest to the Ovac-site produces two σ-type bonds, leading to the emergence of a new localized mid-gap state. This hybrid 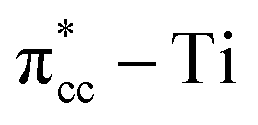 3d defect state can account for the ∼0.7 eV decrease in the band-gap observed in previous optical measurements of carbon-coated TiO2, formed after TiO2 exposure to C2H4 gas (Mater. Lett. 108, 2013, 134). Subsequent calculations on two initial decomposition pathways of C2H4 adsorbed on the Ovac-site show that the C–H bond and C–C bond cleaving require high activation barriers where the C–H bond cleaving is slightly easier compared to the C–C bond cleaving, 2.94 eV and 3.01 eV, respectively, (as calculated using DFT-D2+Ud = 3 eV) and the final states of the two initial decomposition pathways show similar endothermic characteristics. This finding indicates that the surface Ovac-sites tend to favor the formation of molecularly adsorbed Ti-bound sp3-C2H4 (–Ti–CH2CH2–Ti–) as compared to the dissociative adsorption case.
3d defect state can account for the ∼0.7 eV decrease in the band-gap observed in previous optical measurements of carbon-coated TiO2, formed after TiO2 exposure to C2H4 gas (Mater. Lett. 108, 2013, 134). Subsequent calculations on two initial decomposition pathways of C2H4 adsorbed on the Ovac-site show that the C–H bond and C–C bond cleaving require high activation barriers where the C–H bond cleaving is slightly easier compared to the C–C bond cleaving, 2.94 eV and 3.01 eV, respectively, (as calculated using DFT-D2+Ud = 3 eV) and the final states of the two initial decomposition pathways show similar endothermic characteristics. This finding indicates that the surface Ovac-sites tend to favor the formation of molecularly adsorbed Ti-bound sp3-C2H4 (–Ti–CH2CH2–Ti–) as compared to the dissociative adsorption case.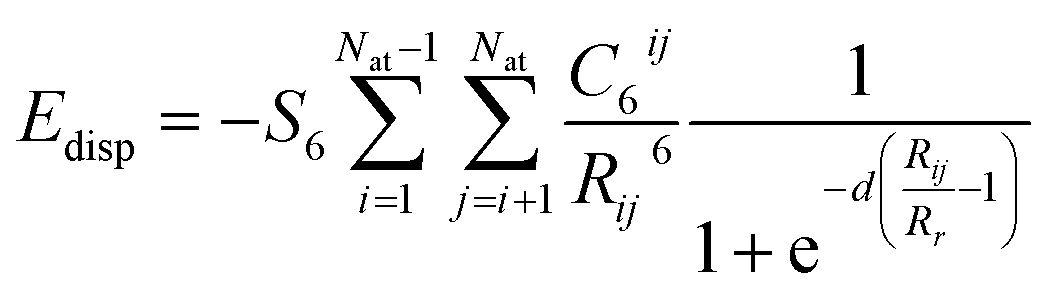
 of C2H4. These two frontier molecular orbitals (MOs) often play a significant part in C2H4 interaction with various solid surfaces. By observing the plot of the orbital-resolved LDOS of C2H4 adsorbed on the Ovac site (Fig. 5), it can be seen that there is a strong hybridization between the p-states of C2H4 C–C with the Ti 3d-states, i.e., two adjacent Ti atoms in the vicinity of the Ovac.65 Another noticeable feature is the emergence of a localized occupied state below the Fermi level, which formed by the mixing of the
of C2H4. These two frontier molecular orbitals (MOs) often play a significant part in C2H4 interaction with various solid surfaces. By observing the plot of the orbital-resolved LDOS of C2H4 adsorbed on the Ovac site (Fig. 5), it can be seen that there is a strong hybridization between the p-states of C2H4 C–C with the Ti 3d-states, i.e., two adjacent Ti atoms in the vicinity of the Ovac.65 Another noticeable feature is the emergence of a localized occupied state below the Fermi level, which formed by the mixing of the 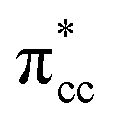 of C2H4 with the occupied Ti 3d defect states.66 This hybridization implies that charges from occupied defect states of Ti 3d are transferred to the
of C2H4 with the occupied Ti 3d defect states.66 This hybridization implies that charges from occupied defect states of Ti 3d are transferred to the 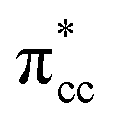 of C2H4, indicating a back donation from the defect states to C2H4 that subsequently weakened the adsorbed C2H4 C–C bond. The weakening because of the depletion of charges in the C–C region upon adsorption can also be observed, whereas accumulation of charges took place in the two C–Ti bonding and C–H bonding regions (Fig. 5d). The accumulation of charges in the C–H bonding regions subsequently tilted the four hydrogen atoms, and as a consequence the C2H4 minimized the repulsive interactions between its bonding electrons. Fig. 5 also shows the plot of the spin density profile within the energy range of Ti 3d defect states –
of C2H4, indicating a back donation from the defect states to C2H4 that subsequently weakened the adsorbed C2H4 C–C bond. The weakening because of the depletion of charges in the C–C region upon adsorption can also be observed, whereas accumulation of charges took place in the two C–Ti bonding and C–H bonding regions (Fig. 5d). The accumulation of charges in the C–H bonding regions subsequently tilted the four hydrogen atoms, and as a consequence the C2H4 minimized the repulsive interactions between its bonding electrons. Fig. 5 also shows the plot of the spin density profile within the energy range of Ti 3d defect states – 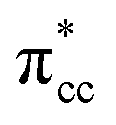 derived-bonding localized mid-gap state that shows two σ-type bonding profiles between both C atoms of C2H4 with the two Ti atoms near the vacancy site. These two σ-type interactions (or the so-called di-σ complex) characterize the rehybridization of C2H4 into the sp3-type upon its adsorption on the Ovac site. It is interesting to note that previous experimental work has shown that coating a anatase TiO2 surface using C2H4 and acetylene as carbon sources resulted in the decrease of the TiO2 band-gap by about ∼0.7 eV, subsequently increasing the photoactivity of the C-modified TiO2 under visible light.22 These present results clarified at least one possible origin of this band-gap decrement. The hybrid
derived-bonding localized mid-gap state that shows two σ-type bonding profiles between both C atoms of C2H4 with the two Ti atoms near the vacancy site. These two σ-type interactions (or the so-called di-σ complex) characterize the rehybridization of C2H4 into the sp3-type upon its adsorption on the Ovac site. It is interesting to note that previous experimental work has shown that coating a anatase TiO2 surface using C2H4 and acetylene as carbon sources resulted in the decrease of the TiO2 band-gap by about ∼0.7 eV, subsequently increasing the photoactivity of the C-modified TiO2 under visible light.22 These present results clarified at least one possible origin of this band-gap decrement. The hybrid 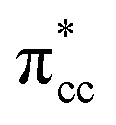 – 3d defect state resulting from C2H4 – reduced anatase TiO2 (001) coupling (located at ∼1.0 eV below the conduction band minimum) can account for the increasing photoactivity of C-modified TiO2, thus extending the system photoactivity to the visible light range. Thus, exposing the system to ultraviolet-visible light can trigger electronic excitation from the hybrid
– 3d defect state resulting from C2H4 – reduced anatase TiO2 (001) coupling (located at ∼1.0 eV below the conduction band minimum) can account for the increasing photoactivity of C-modified TiO2, thus extending the system photoactivity to the visible light range. Thus, exposing the system to ultraviolet-visible light can trigger electronic excitation from the hybrid 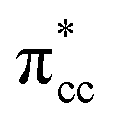 – 3d defect state to the conduction band that accounts for the experimentally observed decreasing band-gap. This is similar to other electron-acceptor molecules adsorbed on a reduced TiO2 surface such as oxygen (O2), where a localized mid-gap state was also observed upon adsorption.67 In addition, a hybrid-DFT calculation was also performed to check the consistency of the present result (C2H4 adsorbed on the Ovac-site) obtained by DFT+Ud (Ud = 3 eV). The optimized geometry and adsorption energy results obtained using hybrid-DFT are in good agreement with the results obtained from DFT+Ud (Ud = 3 eV) (Table 3). This consistency ensures the reliability of the chosen Ud value. To this end, it can be concluded that excess electrons because of the oxygen vacancy play a vital role to stabilize C2H4 adsorption on the reduced anatase TiO2 (001) surface. The present finding is in contrast to the C2H4 adsorption on clean/pristine anatase TiO2 (001), in which C2H4 adsorption is mainly driven by the dispersion (or vdW) interaction.15
– 3d defect state to the conduction band that accounts for the experimentally observed decreasing band-gap. This is similar to other electron-acceptor molecules adsorbed on a reduced TiO2 surface such as oxygen (O2), where a localized mid-gap state was also observed upon adsorption.67 In addition, a hybrid-DFT calculation was also performed to check the consistency of the present result (C2H4 adsorbed on the Ovac-site) obtained by DFT+Ud (Ud = 3 eV). The optimized geometry and adsorption energy results obtained using hybrid-DFT are in good agreement with the results obtained from DFT+Ud (Ud = 3 eV) (Table 3). This consistency ensures the reliability of the chosen Ud value. To this end, it can be concluded that excess electrons because of the oxygen vacancy play a vital role to stabilize C2H4 adsorption on the reduced anatase TiO2 (001) surface. The present finding is in contrast to the C2H4 adsorption on clean/pristine anatase TiO2 (001), in which C2H4 adsorption is mainly driven by the dispersion (or vdW) interaction.15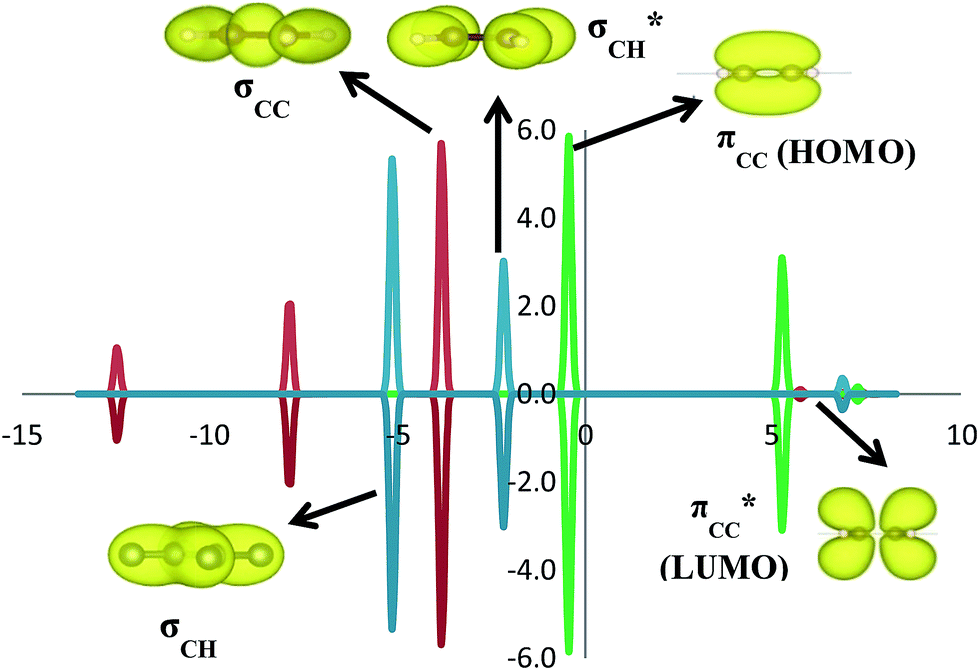
 , (d) charge density difference (CDD) plot of C2H4 adsorbed on Ovac-site. The green color corresponds to the charge depleted region, whereas the yellow corresponds to the charge accumulated region. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3).
, (d) charge density difference (CDD) plot of C2H4 adsorbed on Ovac-site. The green color corresponds to the charge depleted region, whereas the yellow corresponds to the charge accumulated region. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3).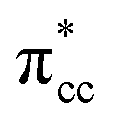 (LUMO). Indeed, some fractions of C2H4
(LUMO). Indeed, some fractions of C2H4 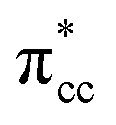 can still be observed within the gap states and also near the valence band edge, showing some electronic filling of the
can still be observed within the gap states and also near the valence band edge, showing some electronic filling of the 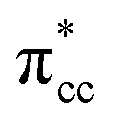 orbital. However, those hybridizations are not strong enough to produce significant stabilization for the C2H4-reduced TiO2 (001) coupling, because most of the broadening of
orbital. However, those hybridizations are not strong enough to produce significant stabilization for the C2H4-reduced TiO2 (001) coupling, because most of the broadening of 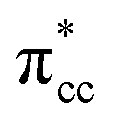 still occupies the region above the Fermi level, resulting in less significant interplay with the πcc (HOMO). This indicates that charge transfer from the surface to the C2H4 is significantly hindered because of the deeper emergence of the defect states in the energy level (or more tightly bound excess electrons) (Fig. 6b). Nevertheless, the tendency of the di-σ bonding formation from the charge density difference (CDD) plot can still be seen, as the charges accumulate in both the C–Ti regions (Fig. 6c). The results obtained from DFT-D2+Ud (Ud = 5 eV) imply another important point that the deeper the energy level of excess electrons (or the more the excess electrons are tightly bound), the more difficult the charge transfer to the C2H4 LUMO will be. This is similar to if the work function of a substrate is controlled, as has also been demonstrated by previous work of O2 adsorption on reduced rutile TiO2 (110) surface.29
still occupies the region above the Fermi level, resulting in less significant interplay with the πcc (HOMO). This indicates that charge transfer from the surface to the C2H4 is significantly hindered because of the deeper emergence of the defect states in the energy level (or more tightly bound excess electrons) (Fig. 6b). Nevertheless, the tendency of the di-σ bonding formation from the charge density difference (CDD) plot can still be seen, as the charges accumulate in both the C–Ti regions (Fig. 6c). The results obtained from DFT-D2+Ud (Ud = 5 eV) imply another important point that the deeper the energy level of excess electrons (or the more the excess electrons are tightly bound), the more difficult the charge transfer to the C2H4 LUMO will be. This is similar to if the work function of a substrate is controlled, as has also been demonstrated by previous work of O2 adsorption on reduced rutile TiO2 (110) surface.29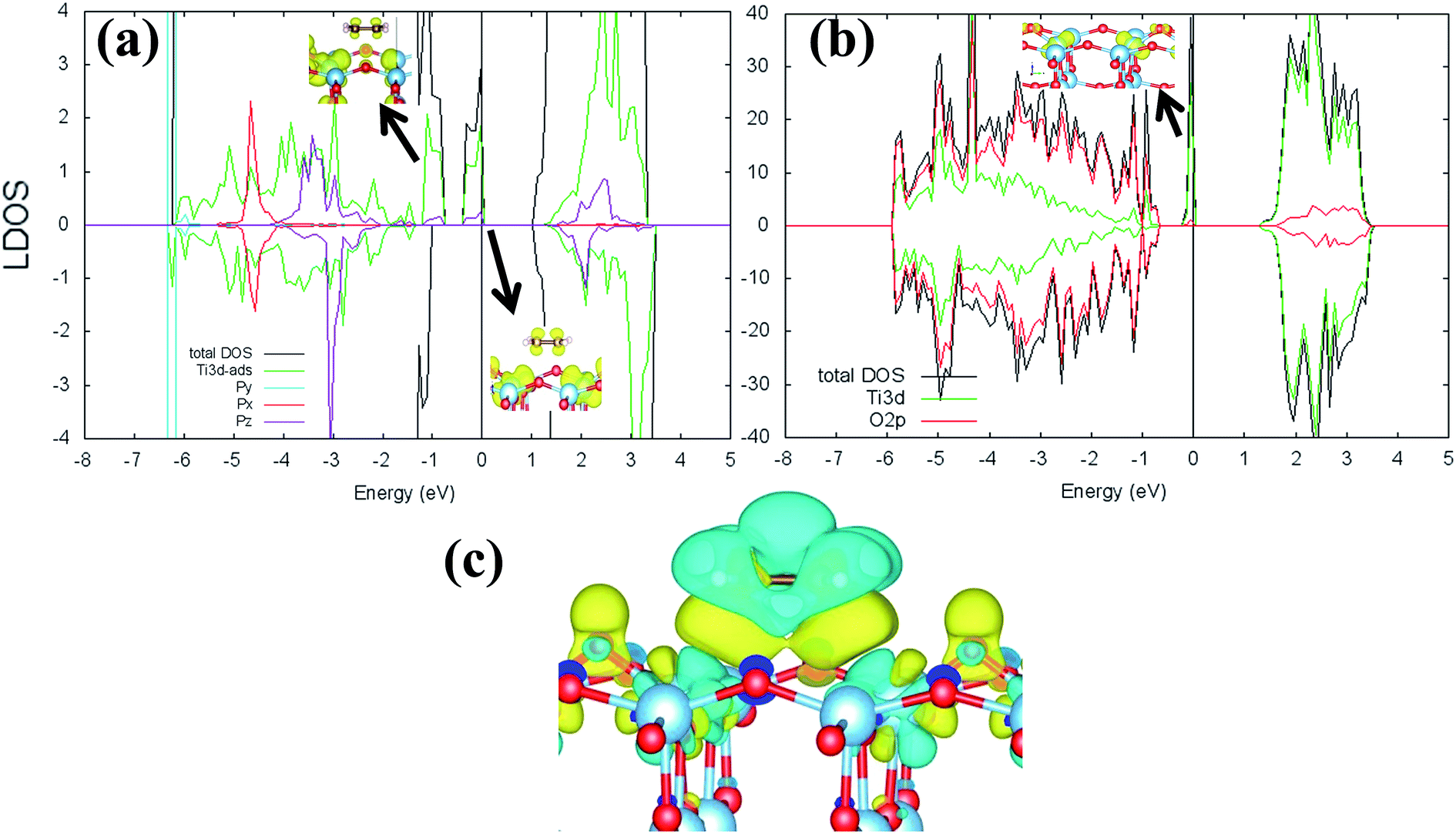
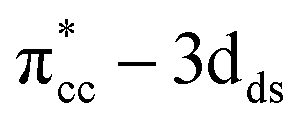 within two energy windows are also shown, (b) LDOS of reduced anatase TiO2 (001) with Ovac calculated at DFT-D2+(Ud = 5 eV), and (c) CDD plot of C2H4 adsorbed on Ovac-site. Green color corresponds to charge depleted region, whereas yellow corresponds to charge accumulated region. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3).
within two energy windows are also shown, (b) LDOS of reduced anatase TiO2 (001) with Ovac calculated at DFT-D2+(Ud = 5 eV), and (c) CDD plot of C2H4 adsorbed on Ovac-site. Green color corresponds to charge depleted region, whereas yellow corresponds to charge accumulated region. The Fermi level is set to 0 eV (isosurface = 0.02 e Å−3).