
The synthesis of iminothiophenone-fused quinolines and evaluation of their serendipitous reactions†
Morteza Shiri*a,
Zeinab Faghihia,
Hossein A. Oskoueia,
Majid M. Heravia,
Shima Fazelzadeha and
Behrouz Notashb
aDepartment of Chemistry, Faculty of Physics and Chemistry, Alzahra University, Vanak, Tehran 1993893973, Iran. E-mail: mshiri@alzahra.ac.ir; Fax: +98 21 88041344
bDepartment of Chemistry, Shahid Beheshti University, G. C., Evin, Tehran 1983963113, Iran
First published on 14th September 2016
Abstract
The novel synthesis of tricyclic 2-(cyclohexylimino)thieno[2,3-b]quinolin-3(2H)-ones from the reaction of 2-mercaptoquinoline-3-carbaldehydes and isocyanides in methanol without the use of any additive is described. This protocol proceeds with high atom economy through the formation of C–S and C–C bonds and then oxidation via tandem reaction. Moreover, some other remarkable aspects of this reaction, such as the hydrolysis and three-component reaction with the aromatic amine to yield a highly conjugated Schiff base, are also investigated.
Introduction
The quinolines are widely used as scaffold in a range of synthetic and natural compounds with pharmacological activities.1–5 Antimalarial,6 antibacterial,7 anti-inflammatory,8 antihypertensive,9 hypotensive,10 anti-rheumatic11 and anti-asthmatic properties12 are amongst the many reported compounds. The functionalized quinolines, particularly the heterocycle-fused ones, have also attracted extensive biological and pharmacological consideration.13 The importance of sulfur-containing scaffolds is recognized by synthetic and medicinal chemists to be associated with some interesting bioactivities. Some of them exhibit antagonistic and antioxidant activity, protecting DNA from hazardous free radicals.13,14 The DNA binding, molecular docking, cytotoxic and biological evaluation studies of some Schiff bases derived from the condensation of 2-mercaptoquinoline-3-carbaldehyde with amines have also been reported lately.15,16 There is, therefore, a growing interest to work on the synthesis of novel quinoline derivatives to achieve diverse and sophisticated pharmaceuticals.Due to their divalent carbon atom, the chemistry of isocyanides is different from the rest of the organic structures, in addition to their practically irreversible reactions corresponding to the exothermic conversions of the C(II) into C(IV). Hence, the reactions including isocyanides are more diverse, as a range of reactants can be involved.17 Considering the noticeable reactivity of isocyanides in undertaking facile additions with nucleophiles and electrophiles as well as 2-mercaptoquinoline-3-carbaldehyde 1, with two potent reactive centres comprising the nucleophilic centre of sulfide and carbonyl as an electrophile, the two-component reaction could be of much interest (eqn (1)). Contrary to their interesting structures, the methodology to construct iminothiophenone-fused quinoline 2 scaffolds has not been explored thus far (eqn (1)). Accordingly, the development of convenient synthetic methods to access such heterocycles provided the impetus to examine the two-component reaction between isocyanides and 2-mercaptoquinoline-3-carbaldehyde 1 (eqn (1)). Moreover, the investigation of further plausible reactions other than the main one, including product hydrolysis and a three-component reaction to make diverse products and to elucidate their supplementary aspects, is of much interest and is discussed subsequently.
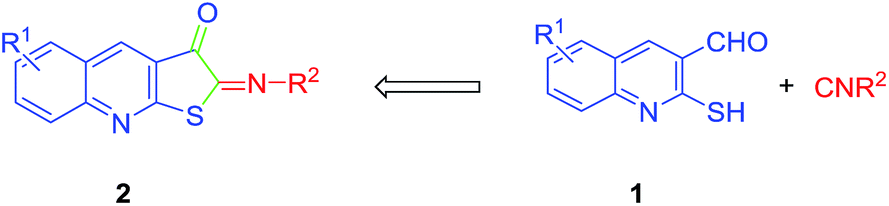 | (1) |
Zahra et al. reported the synthesis of 2-(N-arylhydrazono)-3-oxobenzothiophenes using 2-mercaptobenzoic acid and acylhydrazonoyl chlorides in the presence of catalyst.18 Another structure comprising iminothiophenone fused to a benzene ring was established by Xue et al.19 Adib and co-workers have described a novel method to prepare 2-([2-(alkylimino)-1-benzofuran-3-yliden]amino)benzoic acids.20
On the basis of these reports and our interest in the chemistry of quinoline as well as isocyanides,21–27 we herein report an additive-free synthesis of novel 2-iminothieno[2,3-b]quinolines.
Results and discussion
The typical product, 2-(cyclohexylimino)thieno[2,3-b]quinolin-3(2H)-one 2a, was derived from respective 2-mercaptoquinoline-3-carbaldehyde 1a28,29 and cyclohexylisocyanide by reflux in methanol without using any additive for 2 h to complete the reaction (Scheme 1).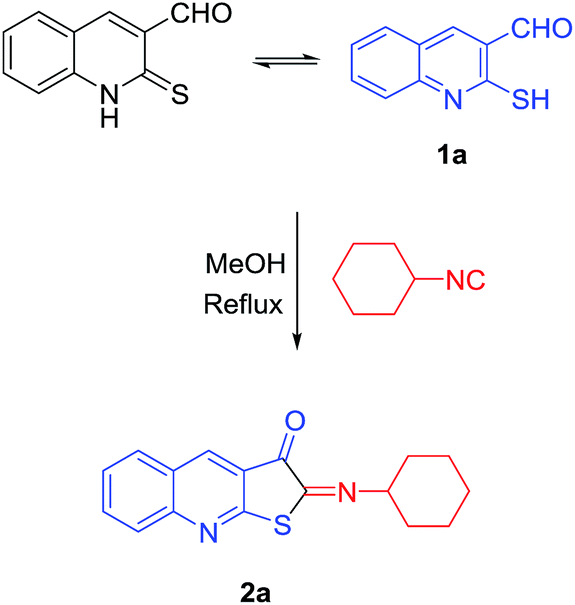 | ||
| Scheme 1 Synthesis of 2-(cyclohexylimino)thieno[2,3-b]quinolin-3(2H)-one 2a. | ||
The structures of the products were elucidated from their IR, 1H-NMR, and 13C-NMR spectra. The IR spectrum of 2a showed absorption bands related to the imine group at 1649 and carbonyl groups at 1710 cm−1. The 1H-NMR spectrum exhibited the characteristic multiplets at δ = 1.25–1.93 for ten protons related to the five CH2 of cyclohexyl and one more cyclohexyl proton, for CH, at 3.35–3.51 ppm. The four aromatic quinoline ring protons appeared at 7.95 to 8.08 ppm as multiplets and the only singlet peak of the quinoline ring at 8.48 ppm. The 13C-NMR spectrum showed 17 signals in agreement with the suggested structure. In the aliphatic region, there are two resonances at 24.4–32.4 and 69.7 ppm for CH2 and CH of cyclohexyl, respectively. The signals related to newly formed bonds, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO, appeared at 153.9 and 184.0 ppm, respectively, and all data verified the molecular structure of 2a.
N and CO, appeared at 153.9 and 184.0 ppm, respectively, and all data verified the molecular structure of 2a.
In optimization study, the same reaction was repeated in different solvents such as CH3CN, CH2Cl2, DMF, DMSO, H2O and EtOH (Table 1). Though the reaction also proceeded in aprotic solvents, better results were achieved in protic solvents. Among protic solvents, methanol yielded the best result. At ambient temperature, the product was formed, but the reaction was not completed without heating. Optimized time for the reaction was 2 h, which yielded 2a in 88%; longer time caused the production of several side products and decreased the product yields.
| Entry | Solvent | Yield | Entry | Solvent | Yield |
|---|---|---|---|---|---|
| 1 | DMF | 10% | 5 | EtOH | 70% |
| 2 | DMSO | 30% | 6 | MeOH | 88% |
| 3 | CH2Cl2 | 40% | 7 | H2O | 40% |
| 4 | CH3CN | 40% | 8 | EtOH/H2O | 60% |
In the next step, the scope of reaction was surveyed. 2-Mercaptoquinoline-3-carbaldehyde 1a–f with different substituents were reacted with cyclohexylisocyanide/tert-butyl isocyanide in refluxing methanol to produce the corresponding iminothiophenone-fused quinolines (Table 2). The reaction of cyclohexylisocyanide was carried out well with quinolines bearing electron-releasing groups such as methyl and methoxy to give 2-(cyclohexylimino)thieno[2,3-b]quinolin-3(2H)-one 2b–d in 82–85% yields (Table 2, entries 2–4). The existence of Cl as electron-withdrawing group led to the product 2e in 73% yield, which is slightly less than others (Table 2, entry 5). Tetracyclic compound 2f was generated in 80% yield, while 2g and 2h, because of using hindered isocyanide in their construction, was formed with 75 and 72% yield (Table 2, entries 6–8).
| Product | 1 | R2 | % | |
|---|---|---|---|---|
| 1 |  |
 |
CycHex | 88 |
| 2 |  |
 |
CycHex | 85 |
| 3 |  |
 |
CycHex | 83 |
| 4 |  |
 |
CycHex | 82 |
| 5 |  |
 |
CycHex | 73 |
| 6 |  |
 |
CycHex | 80 |
| 7 |  |
 |
t-Bu | 75 |
| 8 |  |
 |
t-Bu | 72 |
The reaction of 1i30 as oxygen-containing analogue 1 with cyclohexylisocyanide failed (Scheme 2). It is assumed that the major keto tautomeric form of 2-chloroquinoline-3-carbaldehyde 1i, rather than the enol form, in addition to the lower nucleophilicity of oxygen, were the factors contributing to no product yield (Scheme 2).
 | ||
| Scheme 2 Effect of replacing oxygen with sulfur in heterocyclization with isocyanide. | ||
However, salicylaldehyde 1j and cyclohexylisocyanide under the same condition yielded different products in 50% yield (Scheme 2). Even with changing the solvent and elevating the temperature, better yield was not accessible. Applying these conditions to other salicylaldehyde derivatives gave a mixture of uncharacterizable products.
Mechanism
A plausible mechanism for the formation of 2-(cyclohexylimino)thieno[2,3-b]quinolin-3(2H)-one 2a is considered to be a tandem reaction, as depicted in Scheme 3. This transformation is presumably retriggered by nucleophilic addition of isocyanide to the formyl group in 1a to form A, followed by protonation of oxygen and cyclisation with sulphur atom, giving B, which under dehydrogenation process yielded 2a. To validate the mechanism steps, the reaction was repeated with dried aprotic solvents such as CH3CN, CH2Cl2 and DMF to evaluate the participation of solvent proton in driving the reaction. The reaction progressed in all those solvents, implying that the proton was mainly supplied by 2-mercaptoquinoline-3-carbaldehyde 1 itself. Although the presence of protic solvent noticeably helped and improved the reaction, it was not necessary to use only the protic ones. To verify the oxidation step, the reaction was repeated under N2 atmosphere as well as dry MeOH. No product was practically observed after two hours, indicating that the presence of oxygen was essential for the oxidation step. | ||
| Scheme 3 A plausible mechanism for the formation of 2-(cyclohexylimino)thieno[2,3-b]quinolin-3(2H)-one 2a. | ||
Due to the presence of imine group in the structure of the product 2-(cyclohexylimino)thieno[2,3-b]quinolin-3(2H)-one 2a, it seemed reasonable and interesting to produce the respective ketone when hydrolyzing under mild acidic condition. Accordingly, compound 2a was stirred under 5% at HCl room temperature for 1 hour. The initial imine structure 2a was almost totally converted (Scheme 5). Surprisingly, α-ketoamide 4a was isolated instead of 3. The same process was repeated for 2g as well. As their spectral analysis (such as NMR) revealed afterward, the presence of the signals relating cyclohexyl and tertiobutyl isocyanide protons were solid evidence that hydrolysis had proceeded under a different pathway to afford 4a and 4g, respectively. Basic hydrolysis under 10% NaOH was also examined, which revealed the same result as that in 5% or 10% HCl. The feasible mechanism is shown in Scheme 4.
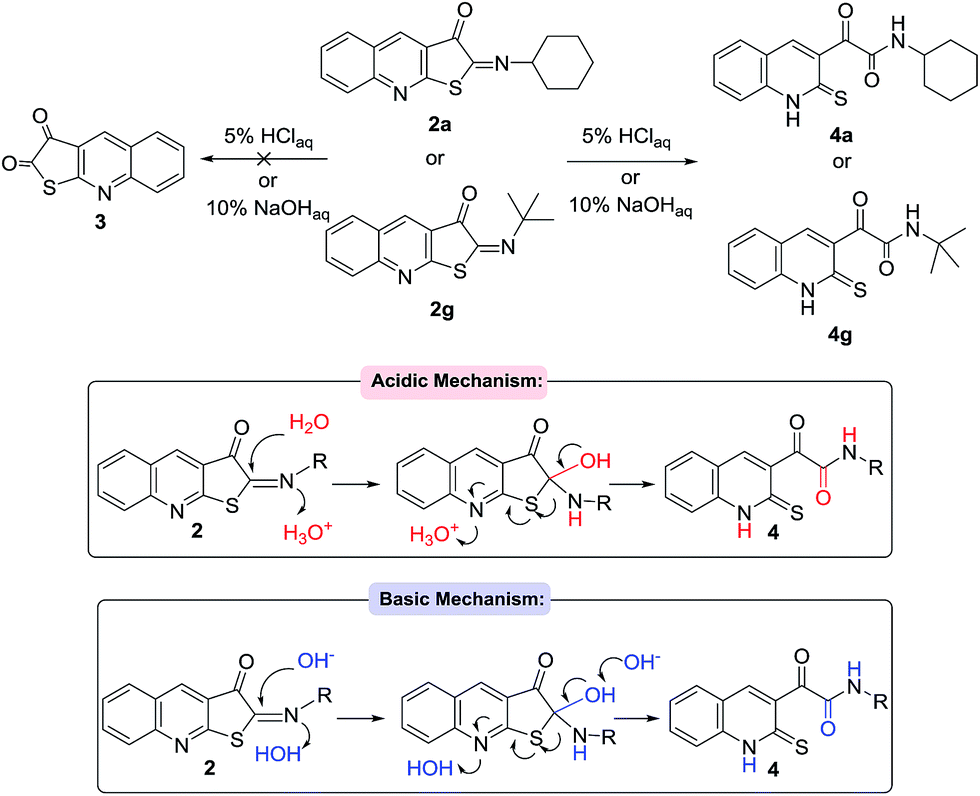 | ||
| Scheme 4 Hydrolysis of compound 2 and probable mechanistic pathways. | ||
The presence of ketone group in the structure of 2-(cyclohexylimino)thieno[2,3-b]quinolin-3(2H)-one 2a encouraged us to also study the possibility of imine formation. The succeeding thought-provoking experiment was to apply a typical aromatic amine to react with 2a via a two-component reaction to form the respective Schiff base 5 (Scheme 5). But the efforts to reach imine 5 using 2a (path A) were in vain. The catalysis-free condition also resulted in no product. The acid catalyst, such as HCl, resulted in hydrolysis product instead of second imine formation (resulting in the same product as 4a), and the amine remained intact. Lewis acid catalysts such as AlCl3 and ZrCl4, in solvents such as CH2Cl2, acetonitrile and methanol under room temperature, also did not obtain any result. Heating also caused the hydrolysis product 4a. Another assumption to reach the imine 5 was trying the one-pot, three-component reaction of 2-mercaptoquinoline-3-carbaldehyde 1a, cyclohexylisocyanides and 4-methyl-aniline.
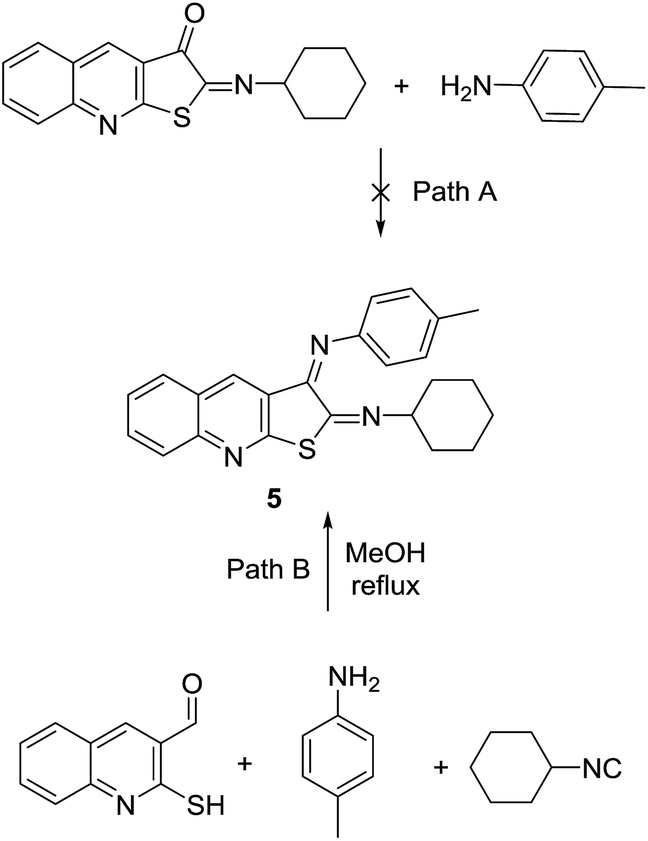 | ||
| Scheme 5 Three-component synthesis of Schiff base 2-(cyclohexylimino)-2,3-dihydro-N-p-tolylthieno[2,3-b]quinolin-3-amine 5. | ||
According to our literature search, the only three-component reaction using amine, isocyanide and salicylaldehyde (as an analogue to mercapto carbaldehyde 1) was reported by Ghandi et al. in 2010. But the oxidation of the cyclic products prevented the formation of the similar structure, imine structure 5, introduced herein.31 In a similar pattern for designing the Ugi reaction, Adib et al. also reported the one-pot reaction between anthranilic acid, salicylaldehyde, and an isocyanide, which unexpectedly afforded a similar product.20
In both reports, the heterocycle containing the exo-imine bond were not formed. Amazingly, in spite of a fruitless two-component reaction (Scheme 5, path A), the three-component reaction applying the starting compounds resulted in the construction of desirable products (Scheme 5, path B). Different conditions to reach this highly conjugated Schiff base were tried. The one-pot reaction at room temperature was not sufficient at all and only yielded minor products. Refluxing without any catalyst produced far better conditions. But the best result was the first treatment of 2-mercaptoquinoline-3-carbaldehyde 1a and 4-methyl-aniline under reflux in methanol to form the initial imine and the addition of the third part (cyclohexylisocyanides) afterward to construct product 5. All in all, the crude product was purified via column chromatography, and shiny red crystalline structure was deduced from NMR spectroscopic data. Lastly, the structure of the product was confirmed by X-ray crystallography of the single crystal 5 (Fig. 1).
 | ||
| Fig. 1 ORTEP diagram for Schiff base 5. | ||
It is assumed that after imine formation, the other sections of the reaction proceed similarly to cyclic products shown in Scheme 3.
Experimental section
Typical experimental procedure for 2a–f
A mixture of 2-mercaptoquinoline-3-carbaldehyde 1a–f29 (0.5 mmol), cyclohexyl or tertiobutyl isocyanides (0.5 mmol) was refluxed in methanol for 1 h, cooled to room temperature and evaporated. The crude solid was purified with 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether:ethylacetate column chromatography to obtain the pure yellow crystalline products 2a–f in good yield (70–88%).
N), 162.68, 184.05 (CO) ppm. Anal. calcd for C17H16N2OS: C, 68.89; H, 5.44; N, 9.45%. Found: C, 68.78; H, 5.53; N, 9.33%.N), 161.68, 184.70 (CO) ppm. Anal. calcd for C18H18N2OS: C, 69.65; H, 5.84; N, 9.02%. Found: C, 69.50; H, 5.92; N, 8.95%.N), 161.63, 184.42 (CO) ppm. Anal. calcd for C18H18N2OS: C, 69.65; H, 5.84; N, 9.02%. Found: C, 69.75; H, 5.92; N, 8.90%.N), 160.33, 184.00 (CO) ppm. Anal. calcd for C18H18N2O2S: C, 66.23; H, 5.56; N, 8.58%. Found: C, 66.11; H, 5.66; N, 8.50%.N), 161.59, 184.73 (CO) ppm. Anal. calcd for C17H15ClN2OS: C, 61.72; H, 4.57; N, 8.47%. Found: C, 61.84; H, 4.64; N, 8.53%.N), 163.28, 184.91 (CO) ppm. Anal. calcd for C21H18N2OS: C, 72.80; H, 5.24; N, 8.09%. Found: C, 72.88; H, 5.32; N, 8.15%.N), 163.03, 184.74 (CO) ppm. Anal. calcd for C15H14N2OS: C, 66.64; H, 5.22; N, 10.36%. Found: C, 66.50; H, 5.39; N, 10.47%.N), 162.32, 185.10 (CO) ppm. Anal. calcd for C16H16N2OS: C, 67.58; H, 5.67; N, 9.85%. Found: C, 67.50; H, 5.77; N, 9.96%.
:1 petroleum ether:ethylacetate column chromatography to obtain the pure yellow crystalline products 2a–f in good yield (70–88%).
N), 162.68, 184.05 (CO) ppm. Anal. calcd for C17H16N2OS: C, 68.89; H, 5.44; N, 9.45%. Found: C, 68.78; H, 5.53; N, 9.33%.N), 161.68, 184.70 (CO) ppm. Anal. calcd for C18H18N2OS: C, 69.65; H, 5.84; N, 9.02%. Found: C, 69.50; H, 5.92; N, 8.95%.N), 161.63, 184.42 (CO) ppm. Anal. calcd for C18H18N2OS: C, 69.65; H, 5.84; N, 9.02%. Found: C, 69.75; H, 5.92; N, 8.90%.N), 160.33, 184.00 (CO) ppm. Anal. calcd for C18H18N2O2S: C, 66.23; H, 5.56; N, 8.58%. Found: C, 66.11; H, 5.66; N, 8.50%.N), 161.59, 184.73 (CO) ppm. Anal. calcd for C17H15ClN2OS: C, 61.72; H, 4.57; N, 8.47%. Found: C, 61.84; H, 4.64; N, 8.53%.N), 163.28, 184.91 (CO) ppm. Anal. calcd for C21H18N2OS: C, 72.80; H, 5.24; N, 8.09%. Found: C, 72.88; H, 5.32; N, 8.15%.N), 163.03, 184.74 (CO) ppm. Anal. calcd for C15H14N2OS: C, 66.64; H, 5.22; N, 10.36%. Found: C, 66.50; H, 5.39; N, 10.47%.N), 162.32, 185.10 (CO) ppm. Anal. calcd for C16H16N2OS: C, 67.58; H, 5.67; N, 9.85%. Found: C, 67.50; H, 5.77; N, 9.96%.Typical experimental procedure for 2j
A mixture of salicylaldehyde (0.5 mmol) and cyclohexylisocyanides (0.5 mmol) was heated at 100 °C under solvent-free condition for 1 h and then cooled to room temperature. The crude solid was purified with 97:3 petroleum ether:ethyl acetate column chromatography to obtain the pure pale yellow crystalline product in 40% yield.
O of amide), 190.07 (CO of ketone) ppm. Anal. calcd for C14H17NO3: C, 68.00; H, 6.93; N, 5.66%. Found: C, 68.10; H, 7.02; N, 5.53%.Typical experimental procedure for 4a–b
The pure compound 2a/2b (0.25 mmol) was vigorously stirred at room temperature in HCl (10%):MeOH (4:1) for an hour. The product extracted with CH2Cl2 was pure and did not need column chromatography. The yellow crystal was formed with high yield of 97%. When using impure compounds 2a/2b as starting materials, column chromatography was needed after work up.
O of ketone), 177.51 (CO of amide), 190.72 (CS) ppm. Anal. calcd for C17H18N2O2S: C, 64.94; H, 5.77; N, 8.91%. Found: C, 65.06; H, 5.89; N, 8.99%.O of ketone), 178.49 (CO of amide), 190.72 (CS) ppm. Anal. calcd for C15H16N2O2S: C, 62.48; H, 5.59; N, 9.71%. Found: C, 62.40; H, 5.68; N, 9.66%.Typical experimental procedure for 5
A mixture of 2-mercaptoquinoline-3-carbaldehyde 1a (0.5 mmol) and 4-methylaniline (0.5 mmol) was refluxed in methanol for about 0.5 h. The cyclohexylisocyanides (0.5 mmol) were then added to the mixture, and reflux was continued for 1 more hour. The solvent was evaporated, and the crude solid was purified with 95:5 petroleum ether:ethyl acetate column chromatography to obtain the pure red crystal 5 in good yield (68%).
N), 160.20, 173 (CN) ppm. Anal. calcd for C24H23N3S: C, 74.77; H, 6.01; N, 10.90%. Found: C, 74.87; H, 6.10; N, 11.05%.Conclusions
In summation, the novel tricyclic structure comprising an iminothiophenone ring fused to quinoline structures was developed in catalyst-free condition. The reaction proceeded via a simple two-component reaction of 2-mercaptoquinoline-3-carbaldehydes and isocyanides. This transformation consists of the formation of three bonds: one C–C and one C–S bond, followed by oxidation, to attain the product in a single synthetic operation. The hydrolysis of the product also resulted in the synthesis of new compounds. Additionally, the typical three-component reaction of aldehyde, amine and isocyanide yielded the amazing crystalline structure, which can be the starting point to synthesize diverse crystalline structures and derivatives.Acknowledgements
We are thankful to Alzahra University and Iran National Science Foundation (INSF) for the financial support.Notes and references
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC.
- J. Marco-Contelles and M. Carreiras, The Friedlander Reaction, Lambert Academic, Saarbrucken, Germany, 2010 Search PubMed.
- J. Marco-Contelles, E. Perez-Mayoral, A. Samadi, M. d. C. Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652–2671 CAS.
- S. Madapa, Z. Tusi and S. Batra, Curr. Org. Chem., 2008, 12, 1116–1183 CAS.
- M. Shiri, M. A. Zolfigol, H. G. Kruger, Z. Tanbakouchian and A. Katritzky, Adv. Heterocycl. Chem., 2011, 185, 139 CrossRef.
- P. Chauhan and S. K. Srivastava, Curr. Med. Chem., 2001, 8, 1535–1542 CrossRef CAS PubMed.
- Y.-L. Chen, K.-C. Fang, J.-Y. Sheu, S.-L. Hsu and C.-C. Tzeng, J. Med. Chem., 2001, 44, 2374–2377 CAS.
- G. Roma, M. Di Braccio, G. Grossi, F. Mattioli and M. Ghia, Eur. J. Med. Chem., 2000, 35, 1021–1035 CrossRef CAS PubMed.
- Y. Morizawa, T. Okazoe, S.-z. Wang, J. Sasaki, H. Ebisu, M. Nishikawa and H. Shinyama, J. Fluorine Chem., 2001, 109, 83–86 CrossRef CAS.
- O. Y. K. Goto and T. Oe, PCT Int. Appl. WO 8401711, A1, 1984.
- Y. Maruyama and M. K. Goto, Terasawa, Ger. Offen. DE 3010751, 1981.
- K. Ukawa, O. Ishiguro and A. Nohara, Chem. Pharm. Bull., 1985, 33, 4432–4437 CrossRef CAS PubMed.
- L. Wu, Y. Wang, H. Song, L. Tang, Z. Zhou and C. Tang, Adv. Synth. Catal., 2013, 355, 1053–1057 CrossRef CAS.
- M. B. Kanani and M. P. Patel, RSC Adv., 2014, 4, 28798–28801 CAS.
- P. Nath and S. D. Dhumwad, Pharma Chem., 2014, 6, 253–261 Search PubMed.
- M. Lokesh, G. Krishnamurthy, H. Bhojyanaik, N. Shashikumar and P. Murali, Pharma Chem., 2014, 6, 192–202 Search PubMed.
- M. Veiderma, Proc. Est. Acad. Sci., 2007, 56, 98 CAS.
- J. A. Zahra, B. A. A. Thaher, M. M. El-Abadelah and R. Boese, Org. Biomol. Chem., 2003, 1, 822–825 CAS.
- J. Xue, Y. Yang and X. Huang, Synlett, 2007, 2007, 1533–1536 CrossRef.
- M. Adib, M. Mahdavi, S. Bagherzadeh, L.-G. Zhu and M. Rahimi-Nasrabadi, Tetrahedron Lett., 2010, 51, 27–29 CrossRef CAS.
- M. Shiri, M. A. Zolfigol, M. Pirveysian, R. Ayazi-Nasrabadi, H. G. Kruger, T. Naicker and I. Mohammadpoor-Baltork, Tetrahedron, 2012, 68, 6059–6064 CAS.
- M. A. Zolfigol, P. Salehi, A. Ghaderi, M. Shiri and Z. Tanbakouchian, J. Mol. Catal. A: Chem., 2006, 259, 253–258 CrossRef CAS.
- M. A. Zolfigol, P. Salehi, A. Ghaderi and M. Shiri, Catal. Commun., 2007, 8, 1214–1218 CrossRef CAS.
- S. Sadjadi and M. M. Heravi, Tetrahedron, 2011, 67, 2707–2752 CrossRef CAS.
- B. Soleymanifard, M. M. Heravi, M. Shiri, M. A. Zolfigol, M. Rafiee, H. G. Kruger, T. Naicker and F. Rasekhmanesh, Tetrahedron Lett., 2012, 53, 3546–3549 CrossRef CAS.
- M. Shiri, B. Farajpour, Z. Bozorgpour-Savadjani, S. A. Shintre, N. A. Koorbanally, H. G. Kruger and B. Notash, Tetrahedron, 2015, 71, 5531–5537 CrossRef CAS.
- M. Shiri, S. Z. Mirpour-Marzoni, Z. Bozorgpour-Savadjani, B. Soleymanifard and H. G. Kruger, Monatsh. Chem., 2014, 145, 1947–1952 CAS.
- A. Srivastava and R. Singh, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2005, 44, 1868 Search PubMed.
- N. J. Parmar, H. A. Barad, B. M. Labana, R. Kant and V. K. Gupta, RSC Adv., 2013, 3, 20719–20725 RSC.
- Z.-C. Liu, B.-D. Wang, B. Li, Q. Wang, Z.-Y. Yang, T.-R. Li and Y. Li, Eur. J. Med. Chem., 2010, 45, 5353–5361 CrossRef CAS PubMed.
- M. Ghandi, P. Asgari, A. Taheri and A. Abbasi, Cent. Eur. J. Chem., 2010, 8, 899–905 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1477633. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra11469e |
| This journal is © The Royal Society of Chemistry 2016 |