
Preparation and characterization of Fe3O4@SiO2@TiO2@Pd and Fe3O4@SiO2@TiO2@Pd–Ag nanocomposites and their utilization in enhanced degradation systems and rapid magnetic separation
Abstract
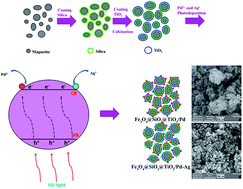
Two new palladium catalysts immobilized on modified magnetic nanoparticles with titanium dioxide shells (Fe3O4@SiO2@TiO2@Pd and Fe3O4@SiO2@TiO2@Pd–Ag nanocomposites) were synthesized and characterized using XRD, SEM, TEM, VSM, EDS, DRS and IR techniques. These catalytic systems showed high activity in the photodegradation of rhodamine B under UV irradiation. The activity of these catalysts was compared and the results showed a higher activity for the Fe3O4@SiO2@TiO2@Pd–Ag nanocomposite due to a synergic effect between silver and palladium. The supported catalysts have the advantage of being completely recoverable with the simple application of an external magnetic field.
 Please wait while we load your content...
Please wait while we load your content...

