
Simultaneous determination of guanine and adenine in the presence of uric acid by a poly(para toluene sulfonic acid) mediated electrochemical sensor in alkaline medium†
Abstract
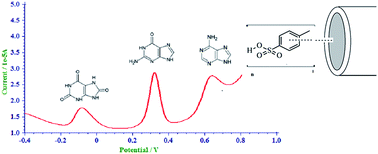
Rapid and sensitive determination of purine bases is vital in clinical analysis. An electrochemical sensor for the determination of the purine bases, guanine, adenine and uric acid, fabricated using a glassy carbon electrode modified with poly(para toluene sulfonic acid), is reported here. In the square wave mode, the modified glassy carbon electrode was able to produce well defined and well separated oxidation peaks for guanine, adenine and uric acid with 0.1 M sodium hydroxide as the supporting electrolyte. The oxidation peak currents for guanine and adenine, showed a dynamic range from 10–100 μM and 20–800 μM with a limit of detection of 0.35 μM and 0.78 μM respectively when determined simultaneously in the presence of uric acid. In simultaneous determination, uric acid also showed a linear increase of oxidation peak current in the concentration range of 10–100 μM, with a limit of detection of 5.88 μM. The variation of peak parameters with scan rate was studied to determine the nature of electro-oxidation and the number of electrons involved in the electrode process. The simultaneous determination of guanine, adenine and uric acid in acid denatured Herring sperm using the fabricated sensor is described. The excellent results obtained indicate that the sensor can be applied for the simultaneous as well as individual determination of guanine, adenine and uric acid in real samples.
 Please wait while we load your content...
Please wait while we load your content...

