
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInorganic–organic hybrid materials based on PbBr2 and pyridine–hydrazone blocks – structural and theoretical study†
Ghodrat
Mahmoudi
*a,
Vladimir
Stilinović
*b,
Antonio
Bauzá
c,
Antonio
Frontera
 *c,
Agata
Bartyzel
d,
Catalina
Ruiz-Pérez
e and
Alexander M.
Kirillov
*f
*c,
Agata
Bartyzel
d,
Catalina
Ruiz-Pérez
e and
Alexander M.
Kirillov
*f
aDepartment of Chemistry, Faculty of Science, University of Maragheh, P.O. Box 55181-83111, Maragheh, Iran. E-mail: mahmoudi_ghodrat@yahoo.co.uk
bDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, HR-10000 Zagreb, Croatia. E-mail: vstilinovic@chem.pmf.hr
cDepartment of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain
dDepartment of General and Coordination Chemistry, Maria Curie-Skłodowska University, Sq. 2, 20-031 Lublin, Poland
eLaboratorio de Rayos X y Materiales Moleculares (MATMOL), Departamento de Física, Facultad de Ciencias (Sección Fisica), Universidad de La Laguna, E-38204 La Laguna, Tenerife, Spain
fCentro de Química Estrutural, Complexo I, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. E-mail: kirillov@tecnico.ulisboa.pt
First published on 15th June 2016
Abstract
Five lead(II) coordination compounds based on PbBr2 and a series of neutral hydrazone and hydrazine ligands (L1–L5) were prepared and structurally characterised, namely [Pb(μ2-Br)(Br)(L1)]2 (1), [Pb(μ2-Br)(Br)(μ2-L2)]n (2), [Pb(μ2-Br)(Br)(μ3-L3)]n (3), [Pb(μ2-Br)(Br)(μ2-L4)]n (4) and [Pb3(μ3-Br)2(μ2-Br)4(L5)2]n (5). In all compounds, there are bridging bromide ligands that interconnect Pb(II) centres and generate either [PbBr2]2 dimers (in 1, 2 and 3) or [PbBr2]n chain motifs (in 4) and [Pb3Br6]n ribbons (in 5). These correspond to three structural fragments present in the lead(II) bromide structure. Depending on the terminal (in 1 and 5) or μ2- and μ3-bridging (in 2, 3 and 4) coordination modes of organic building blocks, the [PbBr2]n fragments constitute discrete molecules (1) or extend to structurally distinct 1D (2 and 5) or 2D (3 and 4) metal–organic networks. Topological analysis and classification of these networks in 2–5 were performed, disclosing underlying chains or layers with the 2C1, 3,4L83, hcb topologies, and a trinodal 3,4,6-connected net of unprecedented topology, respectively. Theoretical calculations (DFT) were employed to analyze some relevant noncovalent interactions observed in the solid state. In particular the inter-ligand π–π stacking interactions in 1 and the influence of the metal coordination on their strength were analyzed. In 3, the role of intramolecular tetrel and π–hole unconventional interactions in the solid state architecture was demonstrated.
1. Introduction
Coordination polymers (CPs) represent nowadays one of the most explored types of compounds in the areas of crystal engineering, coordination and materials chemistry. This is primarily governed by a relatively easy adjustment of the combination of metal nodes and organic building blocks, which opens enormous possibilities towards the fabrication of different materials with various structures, topologies and functional properties.1 Depending on their composition and properties, CPs can have potential applications in the fields of gas adsorption/storage, separation, catalysis, sensing and magnetism.2–6 Since the structural features and functional properties of CPs frequently depend on the coordination modes of metal centres, type of ligands and presence of supramolecular interactions, the selection of organic building blocks plays an important role.7–12 If the organic ligands are neutral molecules, coordination polymers typically bear (inorganic) counter-ions to equalize charges. However, in many cases these counter-ions can act as additional (monodentate and/or bridging) ligands and significantly alter the resulting structures of CPs.13–19In particular, coordination polymers of lead(II) have recently received an increasing attention because of specific and very versatile coordination behaviour of this metal as well as unique supramolecular features and interesting physical properties of such compounds.20–28 In fact, various Pb(II) CPs have found potential applications, for example, as luminescent,29–34 ion exchanging35 and optical materials.36 Since lead(II) ions have an affinity to organic ligands containing O, N and S donor atoms,37–41 various hydrazone building blocks are often used for assembling Pb(II) CPs. On the other hand, the interest in lead(II) coordination polymers also arises from their rich family of halometalates – compounds derived from lead(II) halides and organic ligands.42,43 These represent particularly interesting examples as many of them can be described as inorganic–organic hybrid materials44–47 due to interconnections realised solely by Pb–X bonds in some directions. Bearing these points in mind, herein we report the synthesis, characterization, crystal structures, topologies and theoretical investigation of five coordination compounds 1–5 derived from lead(II) bromide and five pyridine–hydrazone ligands (L1–L5; Scheme 1). The obtained products vary from a discrete dimer [Pb(μ2-Br)(Br)(L1)]2 (1) to 1D coordination polymers [Pb(μ2-Br)(Br)(μ2-L2)]n (2) and [Pb3(μ3-Br)2(μ2-Br)4(L5)2]n (5), and 2D metal–organic networks [Pb(μ2-Br)(Br)(μ3-L3)]n (3) and [Pb(μ2-Br)(Br)(μ2-L4)]n (4). We also demonstrate how an alteration of the orientations of terminal pyridine groups and the bulkiness of the ligand may control the dimensionality and topology of the metal–organic network. Furthermore, a remarkable feature of all the obtained compounds consists of the presence of the PbBr2 subunits interconnected into the [PbBr2]n fragments, which correspond to motifs of the PbBr2 precursor structure48 (Fig. 1). The sizes and dimensionalities of these inorganic fragments are also controlled by the choice of the organic ligand.
 | ||
| Scheme 1 Structural diagrams of the employed ligands (L1–L5). | ||
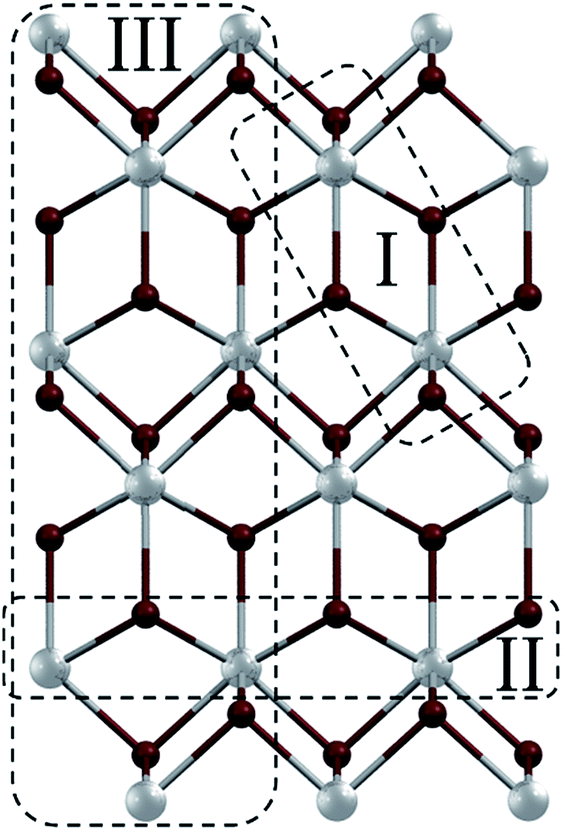 | ||
| Fig. 1 A [101] layer in the structure of lead(II) bromide48 with marked [PbBr2]n fragments I–III that can be identified in the structures of 1–5: fragment I – [PbBr2]2 dimer, fragment II – [PbBr2]n chain, and fragment III – [PbBr2]n triple chain. | ||
2. Experimental
2.1. Materials and measurements
All ligands were prepared following the reported method as described elsewhere.49 All other reagents and solvents were commercially available and used as without further purification. FT-IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer. Microanalyses were performed using a Heraeus CHN–O-Rapid analyser. Melting points were measured on an Electrothermal 9100 apparatus.Caution! Lead and its compounds are toxic.41 Only a small amount of these materials should be prepared and handled with care.
2.2. Synthesis and analytical data of compounds 1–5
Synthesis was performed in a branched tube apparatus. PbBr2 (5 mmol) and the corresponding ligand L1–L5 (5 mmol) were placed in the main arm of a branched tube. Methanol (15 mL) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature. Crystals of products 1–5 were formed in 5 days in the cooler arm and were filtered off, washed with acetone and ether, and dried in air.2.3. X-ray crystallography
Single crystals of 1–5 suitable for X-ray analyses were selected and crystallographic data were collected at 100(2) K on a Bruker AXS SMART APEX CCD diffractometer using Mo Kα radiation (λ = 0.71073 Å) in the ω-scan mode. The detector frames were integrated by use of the SAINT50 program and the empirical absorption corrections were performed using SADABS program.51 All the structures were solved by direct methods and refined by full matrix least-squares procedures using the SHELXTL.52 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms bonded to carbon atoms were placed in calculated positions with isotropic U values 1.2 times higher than those of the parent atom. The hydrogen atoms bonded to nitrogen have been located from the electron difference map and refined isotropically, with the exception of 5 where the hydrogen atom was placed in the calculated position. Materials for publication were prepared using SHELXTL and PLATON.53 Details of crystallographic data are given in Table 1.| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Chemical formula | C54H48N8Pb2Br4 | C12H10N4OPbBr2 | C13H12N4OPbBr2 | C13H12N4OPbBr2 | C26H24N8O2Pb3Br6 |
| M r | 1543.00 | 593.25 | 607.28 | 607.28 | 1581.56 |
| Crystal system | Triclinic | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/c | P21/n |
| a/Å | 10.1876(4) | 7.5893(3) | 8.2036(11) | 9.3608(3) | 7.50290(10) |
| b/Å | 10.6582(4) | 10.6608(5) | 8.7101(12) | 16.1741(6) | 17.6124(2) |
| c/Å | 12.8044(5) | 10.7299(9) | 10.9517(15) | 10.6812(4) | 14.2437(2) |
| α/° | 94.099(1) | 97.040(5) | 100.390(3) | 90 | 90 |
| β/° | 103.293(1) | 102.644(5) | 92.092(3) | 99.959(3) | 101.0623(7) |
| γ/° | 105.080(2) | 99.802(5) | 94.892(3) | 90 | 90 |
| V/Å3 | 1293.66(9) | 823.10(9) | 765.83(18) | 1592.79(10) | 1847.25(4) |
| Z | 1 | 2 | 2 | 4 | 2 |
| ρ calc/(g cm−3) | 1.981 | 2.394 | 2.633 | 2.532 | 2.843 |
| μ/mm−1 | 9.632 | 15.103 | 16.235 | 15.612 | 20.164 |
| F[000] | 732 | 540 | 556 | 1112 | 1416 |
| Crystal size/mm3 | 0.51 × 0.39 × 0.18 | 0.31 × 0.21 × 0.09 | 0.14 × 0.06 × 0.06 | 0.25 × 0.20 × 0.18 | 0.16 × 0.14 × 0.05 |
| T/K | 100(2) | 293(2) | 110(2) | 293(2) | 103(2) |
| Reflections collected | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 494 494 |
13960 |
16558 |
6714 | 42758 |
| Unique reflections | 8524 | 3948 | 5837 | 3494 | 5384 |
| Observed reflections | 6865 | 3570 | 5274 | 2517 | 4571 |
| Parameters | 307 | 169 | 190 | 191 | 206 |
| R 1(obs) | 0.0252 | 0.0398 | 0.0228 | 0.0436 | 0.0206 |
| wR2(all) | 0.0614 | 0.1074 | 0.0518 | 0.0853 | 0.0415 |
| S | 1.024 | 1.070 | 1.028 | 0.993 | 0.985 |
| Max./min Δρ/(e Å−3) | 1.865/−1.744 | 2.789/−1.276 | 1.252/−1.739 | 1.354/−1.242 | 0.963/−0.848 |
2.4. Topological analysis
Topological analysis and classification of metal–organic networks in 2–5 were carried out using Topos software and following the concept of the simplified underlying net.54,55 Such nets were obtained by reducing all ligands to the respective centroids and maintaining their connectivity by coordination bonds. Terminal bromide ligands were also omitted.2.5. Theoretical methods
All calculations were carried out using the TURBOMOLE version 7.0 using the BP86-D3/def2-TZVP level of theory.56 To evaluate the interactions in the solid state, we used the atom coordinates as obtained from the crystal structures. This procedure and level of theory were successfully used to evaluate similar interactions.57 The interaction energies were computed by calculating the difference between the energies of isolated monomers and their assembly. The interaction energies were corrected for the Basis Set Superposition Error (BSSE) using the counterpoise method.58 The molecular electrostatic potential (MEP) surfaces were computed at the B3LYP/6-31+G* level of theory by means of the Spartan software.59 The “atoms-in-molecules” (AIM)60 analysis was performed at the BP86-D3/def2-TZVP level of theory. The calculation of AIM properties was done using the AIMAll program.613. Results and discussion
One-pot synthesis in a branched tube apparatus and using as reagents lead(II) bromide and different organic building blocks L1–L5 in MeOH afforded a new series of lead(II) crystalline products 1–5. They range from a discrete dilead(II) complex [Pb(μ2-Br)(Br)(L1)]2 (1) to 1D coordination polymers [Pb(μ2-Br)Br(μ2-L2)]n (2) and [Pb3(μ3-Br)2(μ2-Br)4(L5)2]n (5), and 2D metal–organic networks [Pb(μ2-Br)(Br)(μ3-L3)]n (3) and [Pb(μ2-Br)(Br)(μ2-L4)]n (4), depending on the coordination modes of hydrazone and bromide ligands. All compounds were obtained in good yields, and were fully characterized by elemental analysis, FTIR spectroscopy, single crystal X-ray diffraction, topological analysis and theoretical calculations.3.1. Crystal structures of 1–5
In all five crystal structures there are discernible fragments of the PbBr2 structure. In three structures (1, 2 and 3), the PbBr2 blocks are connected into centrosymmetric dimeric motifs (fragment I), in 4 their interconnection is in a head-to-tail fashion leading to chains (fragment II), while in 5 the largest ribbon-like fragment of the PbBr2 structure (fragment III) has been preserved.Although compounds 1, 2 and 3 all comprise dilead(II) blocks [Pb2(μ2-Br)2(Br)2], their overall structures are markedly different due to the distinct coordination modes of hydrazone ligands L1–L3 that act either as terminal ligands (L1; 1) or μ2- (L2; 2) and μ3-spacers (L3; 3). Thus, the structures of 1–3 vary from a 0D dimer 1 to a 1D coordination polymer 2 and a 2D metal–organic network 3.
In 1, the seven-coordinate Pb1 atom is surrounded by four nitrogen atoms of L1, one terminal bromide (Br2) and two bridging bromide ligands (Br1), forming a [Pb(μ2-Br)(Br)(L1)]2 dimer (Fig. 2). The dimers are centrosymmetric (they lie on crystallographic inversion centres) so that the two bridging bromide ligands and the two Pb1 atoms are related by an inversion centre. The bridging bromide ligands are almost equidistant from the two Pb centres [Pb1–Br1 3.0541(3), Br1–Pb1′ 3.0735(4) Å], while the binding of the terminal Br2 ligand shows a shorter distance [Pb1–Br2 2.9228(4) Å]. A considerable bulkiness of L1 and its N,N,N,N-chelating mode prevent an additional binding to adjacent Pb centres, thus resulting in a discrete structure. The neighbouring dimers are however interconnected by weak interactions. The most significant intermolecular contacts are C–H⋯Br hydrogen bonds, with the terminal bromide acting as an acceptor in two such contacts [C2–H2⋯Br2 of 3.565 Å and C2–H2⋯Br2 of 3.701 Å], and the bridging bromide in one [C22–H22⋯Br1 of 3.550 Å].
 | ||
| Fig. 2 Structural fragments of 1. (top) Centrosymmetric discrete lead(II) dimer. (down) Topological representation of adjacent dimers in the crystal packing pattern (view along the b axis); colour codes: Pb1 atoms (grey balls), μ2-Br linkers (brown), centroids of terminal L1 ligands (cyan). | ||
In a 1D coordination polymer 2 (Fig. 3), the Pb1 atom is also seven-coordinate by four atoms from μ2-L2 ligands, one terminal bromide (Br2) and two bridging bromide moieties (Br1). The dimeric [Pb2(μ2-Br)2(Br)2] blocks are quite similar to those in 1, and thus will not be discussed further. However, unlike L1, L2 cannot act as a tetradentate ligand to one lead atom, but rather chelates one Pb(II) atom with two nitrogen (2-pyridyl and hydrazone) and an oxygen atom, while the 3-pyridyl nitrogen atom binds to a neighbouring Pb1 centre. Each Pb1 atom is therefore coordinated by three chelating atoms of one μ2-L2 ligand and a pyridine nitrogen atom of another μ2-L2 moiety. Hence, μ2-L2 acts as a linker between lead(II) dimers interconnecting them into chains along the crystallographic a axis. From the topological viewpoint, the metal–organic chain in 2 can be considered as a 2-connected underlying 1D network with the 2C1 topology (Fig. 3, down). As in 1, there are some hydrogen bonds in 2, namely one N–H⋯Br bond [N3–H3n⋯Br2 3.442 Å] and two weak C–H⋯Br bonds [C6–H6⋯Br2 3.711 Å and C10–H10⋯Br2 3.881 Å], which interconnect the adjacent chains into supramolecular 2D layers perpendicular to the crystallographic c axis.
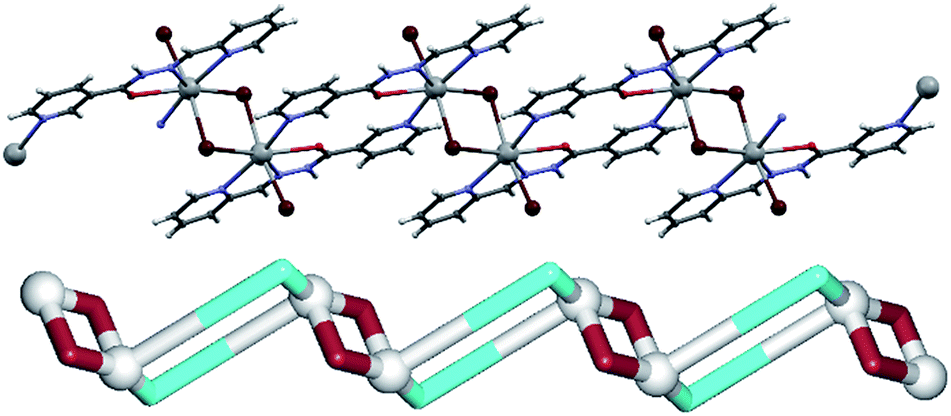 | ||
| Fig. 3 Structural fragments of 2. (top) Metal–organic chain composed of repeating [PbBrL2]2 motifs. (down) Topological representation of a simplified underlying network showing a uninodal 2-connected chain with the 2C1 topology (rotated view along the a axis); colour codes: 2-connected Pb1 nodes (grey balls), centroids of 2-connected μ2-L2 (cyan) and μ2-Br (brown) linkers. | ||
Replacing the 2-pyridyl group with a 3-pyridyl one (L3) makes the hydrazone molecule incapable of chelating to Pb centre with three atoms as was the case in 2, but rather makes L3 acting as a μ3-spacer and thus giving rise to the formation of a 2D coordination polymer structure (Fig. 4). The centrosymmetric [Pb2(μ2-Br)2(Br)2] dimers are still preserved, although their geometry is somewhat different than that observed in 1 and 2. The bridging bromide ligands are not placed equidistantly between the two lead atoms with one Pb1–Br1 bond comparable to those in 1 and 2 [3.0439(4) Å] but the other one [2.8888(4) Å] being shorter than the Pb1–Br1 bond of the terminal bromide [2.9515(5) Å]. The six-coordinate Pb1 atom is surrounded by three bromide ligands as well as two pyridine nitrogen atoms and one carboxyl oxygen from three different μ3-L3 blocks. These interconnect the [Pb2(μ2-Br)2(Br)2] units into sheets perpendicular to the crystallographic a axis. The coordination is hemidirectional which allows the Pb1 centre to participate in an additional weak Pb1⋯N2 contact of 3.228 Å involving the imine nitrogen atom of the O-donating μ3-L3 molecule. The most significant intermolecular contacts are hydrogen bonds involving bromide moieties [N3–H3⋯Br2 3.427 Å and C12–H12⋯Br1 3.585 Å]. Topological analysis of the 2D metal–organic sheet in 3 discloses a binodal 3,4-connected underlying layer with the 3,4L83 topology (Fig. 4, down). This topology is defined by the point symbol of (42·63·8)(42·6), wherein the (42·63·8) and (42·6) notations correspond to the 4-connected Pb1 and 3-connected μ3-L3 nodes, respectively; there are also the 2-connected μ2-Br linkers.
 | ||
| Fig. 4 Structural fragments of 3. (top) 2D metal–organic network composed of [Pb2Br2] motifs and μ3-L3 blocks. (down) Topological representation of a simplified underlying network showing a binodal 3,4-connected layer with the 3,4L83 topology (rotated view along the a axis); colour codes: 4-connected Pb1 nodes (grey balls), centroids of 3-connected μ3-L3 nodes (cyan), 2-connected μ2-Br linkers (brown). | ||
The only difference between the L2 and L4 ligands concerns the presence of a methyl group on the 2-pyridyl side of the molecule. This however leads to quite dramatic changes in the structure of the resulting lead(II) coordination polymer 4 (Fig. 5). In this compound, the Pb1 centre is also seven-coordinate in a similar fashion as in 2 with two bridging and one terminal bromide ligands, three atoms from one μ2-L4 molecule and a 3-pyridyl nitrogen from another one. However, unlike in 2, in the structure of 4 the PbBr2 units are not interconnected into centrosymmetric dimers but rather they bind in a head-to-tail fashion, leading to the generation of [PbBr2]n chain motifs which stretch along the crystallographic c axis. These motifs are then held together by the μ2-L4 linkers, thus giving rise to a 2D metal–organic network (Fig. 5, top). This network can be topologically classified as a uninodal 3-connected underlying layer (Fig. 5, down) with the hcb [Shubnikov hexagonal plane net/(6,3)] topology and the point symbol of (44·62). This layer is assembled from the 3-connected Pb1 nodes and the 2-connected μ2-L4 and μ2-Br linkers.
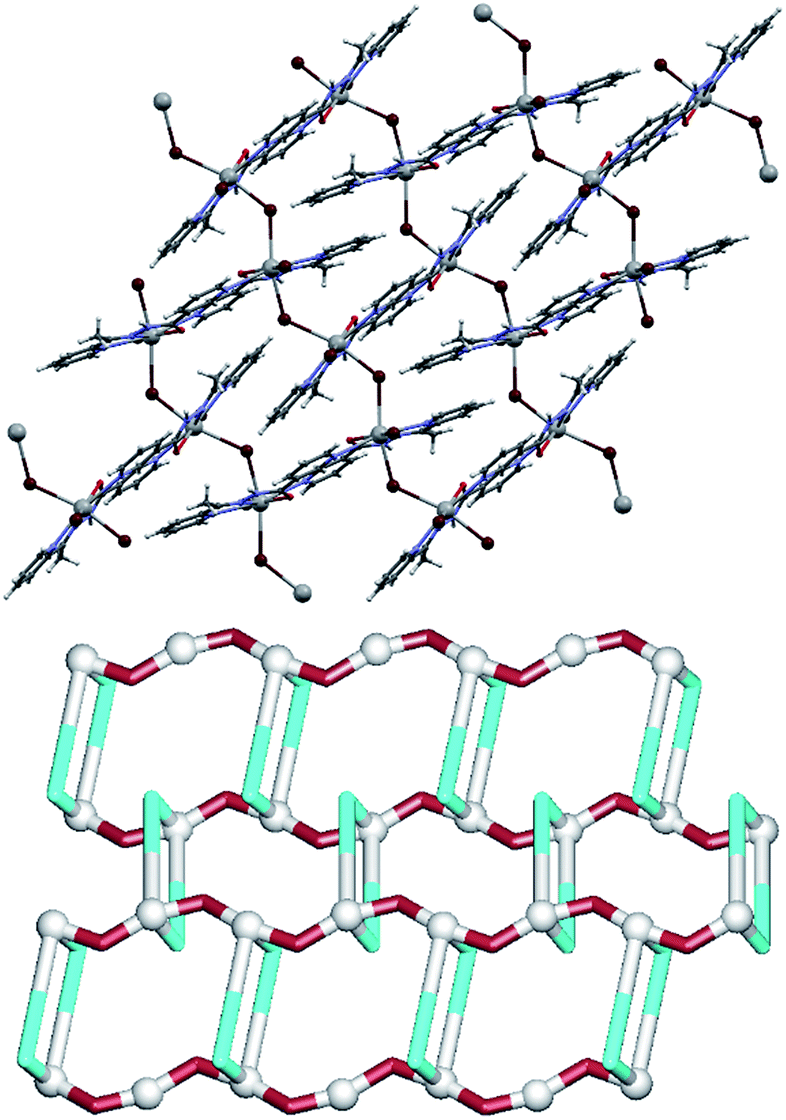 | ||
| Fig. 5 Structural fragments of 4. (top) 2D metal–organic network composed of [Pb(μ2-Br)]n chain motifs interconnected by μ2-L4 linkers. (down) Topological representation of a simplified underlying network showing a uninodal 3-connected layer with the hcb topology (view along the a axis); colour codes: 3-connected Pb1 nodes (grey balls), centroids of 2-connected μ2-L4 linkers (cyan), 2-connected μ2-Br linkers (brown). | ||
In 4, only one μ2-bromide ligand interconnects the adjacent Pb1 centres. The corresponding Pb1–Br bonds are not equal, one is very short [2.8882(10) Å] and the other one is substantially longer [3.1044(10) Å]. The bonding of the terminal bromide ligand is of intermediate length [3.0492(9) Å], which is slightly longer than in the previously described structures. The presence of the methyl group blocks the approach of a hydrogen acceptor to the N–H group of the L4 ligand. Hence, the N–H⋯Br hydrogen bonding described in 2 is not observed in 4. The terminal bromide ligand thus participates only in a very weak hydrogen bond between a methyl group of L4 from a neighbouring layer (C7–H7a⋯Br1 of 3.745 Å), and the bridging bromide with an aromatic hydrogen from the same molecule (C12–H12⋯Br2 of 3.760 Å). The only difference between the L2 and L4 ligands lies in the presence of the methyl group, which in turn causes this difference in hydrogen bonding capability of the ligand. Thus, it appears that the discriminating force between the layered structure with [PbBr2]n chains in 4 and the chain structure with [PbBr2]2 dimers in 2, is the hydrogen bonding between non-covalently bonded structures, i.e. crystal packing forces. This would indicate that there is only a small energy difference between the two structural motifs (fragment I and II).
Compound 5 features the largest fragment of the PbBr2 structure observed within the five studied examples (fragment III). The structure can be described as a metal–organic ribbon with the repeating [Pb3(μ3-Br)2(μ2-Br)4(L5)2]n blocks (Fig. 6). There are two distinct Pb1 and Pb2 centres, one μ3-Br spacer (Br1), two types of μ2-Br linkers (Br2, Br3) and one terminal L5 ligand. The seven-coordinate Pb1 atom is surrounded by four bridging bromide moieties and three atoms of the L3 ligand. The Pb2 atom is six-coordinate and its coordination octahedron is filled solely by the bridging bromide ligands. The Pb–Br distances vary from 2.9775(4) to 3.2218(3) Å and closely correspond to those in the structure of PbBr2. However, unlike in PbBr2 where the bromide ions bridge also to a second layer of lead(II) atoms, in 5 such bonding is blocked by the bulkiness of L5 that acts as a terminal ligand. To get further insight into a very intricate structure of the metal–organic ribbon in 5, we performed its topological analysis. Hence, the ribbon is built from the 6-connected Pb2 and 4-connected Pb1 nodes, 3-connected μ3-Br nodes and the 2-connected μ2-Br linkers (Fig. 6, down). Its topological analysis reveals a trinodal 3,4,6-connected net with the unique topology defined by the point symbol of (32·44)2(32·4)2(34·42), wherein the (32·44), (32·4), and (34·42) indices are those of the Pb1, μ3-Br and Pb2 nodes, respectively. An undocumented type of the present topology was confirmed by a search of different databases.54,55,62,63
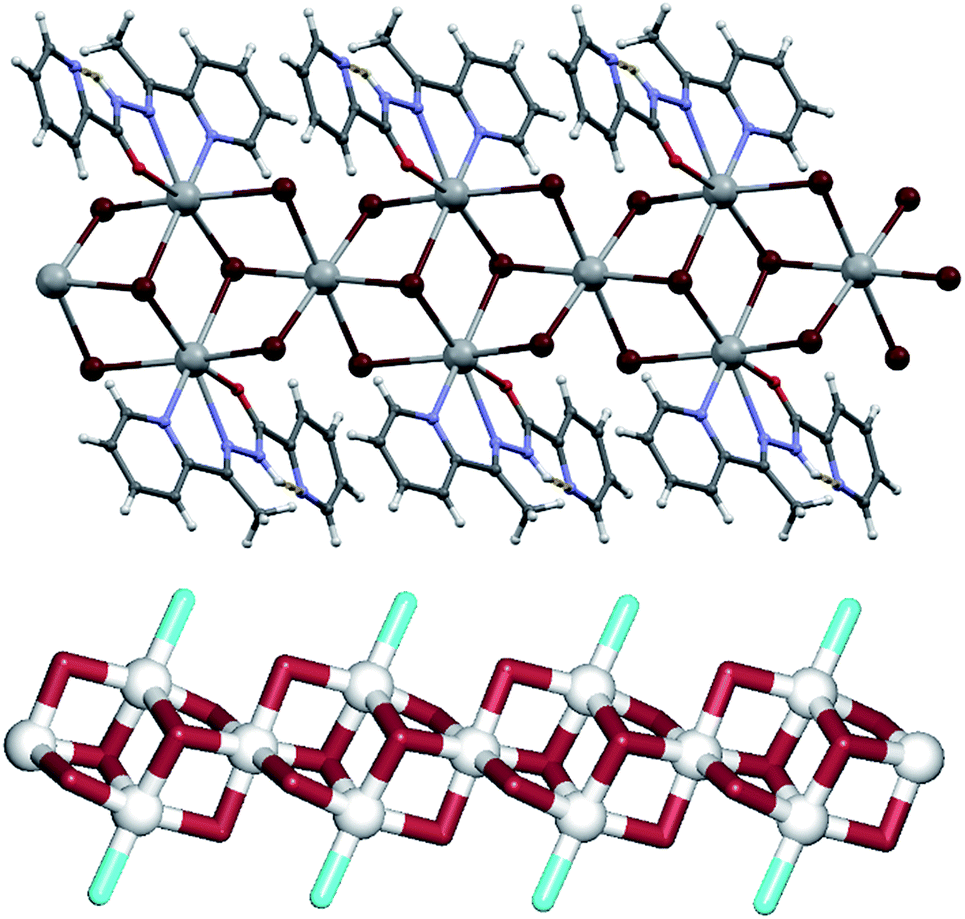 | ||
| Fig. 6 Structural fragments of 5. (top) 1D metal–organic ribbon composed of [Pb3(μ3-Br)2(μ2-Br)4(L5)2]n motifs; it exhibits a fragment III of the PbBr2 structure. Intramolecular N3–H3⋯N4 hydrogen bond is shown as a yellow stippled line. (down) Topological representation of a simplified underlying network showing a trinodal 3,4,6-connected ribbon with the unique topology defined by the point symbol of (32·44)2(32·4)2(34·42) (view along the b axis); colour codes: 6-connected Pb2 and 4-connected Pb1 nodes (grey balls), 3-connected μ3-Br nodes and 2-connected μ2-Br linkers (brown), terminal L5 ligands (cyan). | ||
Further interconnection of the neighbouring [Pb3(μ3-Br)2(μ2-Br)4(L5)2]n ribbons is achieved only through weak hydrogen bonding contacts [C13–H13⋯Br2 3.679 Å and C3–H3a⋯O1 3.225 Å]. Interestingly, the 2-pyridyl nitrogen atom is not involved in the linkage of ribbons neither by bridging to Pb atoms of neighbouring ribbons, nor by acting as a hydrogen donor. This is due to both steric reasons and the presence of an intramolecular hydrogen bond [N3–H3⋯N4 2.627 Å] which, despite being geometrically strained [N3–H3⋯N4 angle of 103°], renders both the amide N–H unable to act as intermolecular hydrogen donor and the 2-pyridyl nitrogen atom as intermolecular H-bond acceptor. This intramolecular hydrogen bond thus makes L5 acting as a tridentate ligand with all available donor atoms chelating to a Pb centre. Therefore, further binding can only be achieved through coordination of additional bromide ions to the Pb atoms. As L5 is only tridentate and sterically much less demanding than L1, in 5 this leads to the generation of the intricate [Pb3(μ3-Br)2(μ2-Br)4(L5)2]n ribbons, while in 1 only the [Pb(μ2-Br)(Br)(L1)]2 dimers are formed.
3.2. Theoretical study
Apart from 1, the solid state architecture of the rest of compounds reported herein is determined by their polymeric nature due to the existence of intricate interconnections of [PbBr2L]2 dimeric units. Regarding the noncovalent interactions and assemblies observed in the solid state architecture of compound 1, we have mainly focused our attention to the π–π stacking interactions involving the aromatic rings of the ligands and the influence of their coordination to the metal centre upon the π–π binding strength.In compound 1, using the crystallographic coordinates, we have evaluated the energetic features of the C–H⋯Br and π–π interactions observed in the solid state, which are important in the crystal packing. In Fig. 7 we show the original structural fragment as obtained by X-ray diffraction, and theoretical models used for the calculations.
 | ||
| Fig. 7 Fragment of the crystal structure (A) and theoretical models (B–D) used to evaluate the H-bonding and π–π stacking interactions compound 1, distances in Å. | ||
As can be observed in Fig. 7A, 1 forms a ladder motif in the solid state due to a combination of weak H-bonds and π-stacking interactions. The C–H⋯Br interaction is established between one aromatic hydrogen atom of L1 and the bromide co-ligand, while the π–π interaction is achieved by the antiparallel stacking of two pyridine moieties of L1. The evaluation of the interaction energy for one dimeric unit retrieved from this infinite layer (see Fig. 7B) shows that this energy is large and negative (ΔE1 = −21.9 kcal mol−1), likely due to enhanced acidity of the H atoms of the ligand due to its coordination to the Pb(II) metal centres and the anionic nature of the electron donor atom (Br), thus favouring the H-bonding interaction and also the antiparallel arrangement of the π-stacking. In an effort to estimate the contribution of each interaction, we have computed an additional theoretical model where the bromine atoms have been replaced by hydrogen (see Fig. 7C) and, consequently, the interaction energy is reduced to ΔE2 = −11.9 kcal mol−1, which is the contribution of the π-stacking and the difference with ΔE1 (−10 kcal mol−1) is the contribution of both H-bonds. The interaction energy for the π-stacking is very large likely due to the antiparallel arrangement and the effect of the metal coordination that increases the dipole–dipole interaction. In an effort to roughly evaluate this contribution, we have computed an additional model where the Pb atoms and counter ions have been eliminated (see Fig. 7D). As a result, the overall interaction energy is significantly reduced to ΔE3 = −4.1 kcal mol−1, confirming the strong influence of the metal coordination on the π-stacking binding energy.
Another aspect that we have analysed theoretically is the noncovalent N⋯Pb interaction observed in 3; it is not present in the related compounds 2, 4 and 5. This is due to the different position of the nitrogen atom of the pyridine moiety relative to the imino group. In compounds 2, 4 and 5 the ligand is tridentate in one side (N,N,O) and monodentate on the other side (see Scheme 1 and Fig. 3, 5 and 6).
However, in 3 the ligand is coordinated to three Pb metal centres, each one in a monodentate manner. Interestingly, one of these monodentate coordination modes is via the O atom of the ligand and this coordination is additionally supported by a noncovalent N⋯Pb tetrel bonding interaction (dashed line, see Fig. 8A). The N⋯Pb distance (3.22 Å) is larger than a normal Pb–N coordination bond but shorter than the sum of van der Waals radii (3.57 Å). We have recently demonstrated64 by combining the CSD analysis and DFT calculations that tetrel bonding interactions are non-isotropic and play an important role in determining the solid state architectures of several Pb(II) metal–organic networks. The presence of this interaction in 3 confirms its importance in the solid state. Moreover, we have also observed a π–hole interaction in this compound between the bromide ligand and the carbon atom of the coordinated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (see blue dashed line in Fig. 8A). It is worth mentioning that this type of noncovalent interactions (counterpart of a σ–hole) can be defined as is a region of positive electrostatic potential that is perpendicular to a portion of a molecular framework. Such interactions have attracted considerable attention in recent years due to their importance in several fields including crystal engineering.65 We have computed the molecular electrostatic potential (MEP) surface of the ligand (see Fig. 8B) where a positive isosurface (blue contour) can be observed. This isosurface reaches the position of the C atom (+14 kcal mol−1) thus explaining this unconventional interaction, which is electrostatically favoured. We further analysed both the tetrel and π–hole interactions using the Bader's theory of “atoms in molecules” that provides an unambiguous definition of chemical bonding.66 We used a fragment of the polymeric structure (see Fig. 8C). The AIM analysis of this fragment confirms the presence of the tetrel bonding interaction which is characterized by the presence of a bond critical point that connects the nitrogen atom of L3 to Pb centre. The π–hole interaction is also characterized by the presence of a bond critical point that connects the bromine atom to the carbon atom of the CO. In both cases the Laplacian of the electron density at the bond critical point is positive, being common for closed shell interactions.66
O (see blue dashed line in Fig. 8A). It is worth mentioning that this type of noncovalent interactions (counterpart of a σ–hole) can be defined as is a region of positive electrostatic potential that is perpendicular to a portion of a molecular framework. Such interactions have attracted considerable attention in recent years due to their importance in several fields including crystal engineering.65 We have computed the molecular electrostatic potential (MEP) surface of the ligand (see Fig. 8B) where a positive isosurface (blue contour) can be observed. This isosurface reaches the position of the C atom (+14 kcal mol−1) thus explaining this unconventional interaction, which is electrostatically favoured. We further analysed both the tetrel and π–hole interactions using the Bader's theory of “atoms in molecules” that provides an unambiguous definition of chemical bonding.66 We used a fragment of the polymeric structure (see Fig. 8C). The AIM analysis of this fragment confirms the presence of the tetrel bonding interaction which is characterized by the presence of a bond critical point that connects the nitrogen atom of L3 to Pb centre. The π–hole interaction is also characterized by the presence of a bond critical point that connects the bromine atom to the carbon atom of the CO. In both cases the Laplacian of the electron density at the bond critical point is positive, being common for closed shell interactions.66
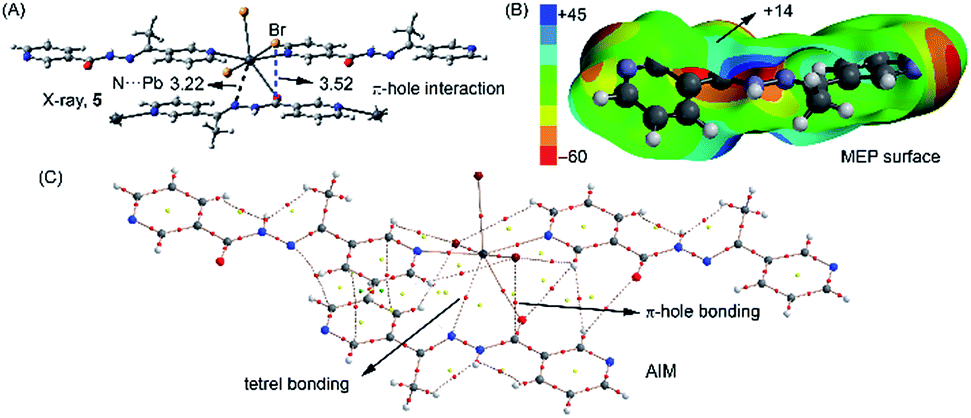 | ||
| Fig. 8 (A) Fragment of the crystal structure of compound 3. Distances in Å. (B) MEP surface of the ligand. Value in kcal mol−1. (C) Distribution of critical points in an fragment of the crystal structure of 3. Bond, ring and cage critical points are represented by red, yellow and green spheres, respectively. The bond paths connecting the bond critical points are also indicated. | ||
4. Conclusions
The present work reported the synthesis, characterization, crystal structures, topological classification and theoretical analysis of a new series of lead(II) coordination compounds driven by bromide and different pyridine–hydrazone building blocks. In the all five compounds there are discernible fragments of the structure of lead(II) bromide. The most common is the smallest fragment – centrosymmetric [Pb2(μ-Br)2(Br)2] dimer. Its presence in three structures (1, 2 and 3) demonstrates the stability of the [PbBr2]2 units as a building block for the generation of metal–organic structures of variable dimensionality, which is achievable through the choice of an additional ligand with a certain potential to act as a linker or spacer.In fact, similar reactions of PbBr2 with L1, L2 or L3 led to discrete dimers 1, 1D (2), or a 2D coordination polymer 3, respectively. Similarly, other structural types of 2D and 1D coordination polymers 4 and 5 were generated from PbBr2 and L4 and L5, respectively. Topological analysis and classification of the simplified underlying networks in the compounds 2–5 were performed, disclosing a uninodal 2-connected chain with the 2C1 topology in 2, a binodal 3,4-connected layer with the 3,4L83 topology in 3, a uninodal 3-connected layer with the hcb topology in 4, and a topologically unique trinodal 3,4,6-connected ribbon in 5.
The variability of the PbBr2 fragments that appear in the structures of coordination polymers 2–5 as a response to rather minor differences in the employed ligands L2–L5 demonstrates a high potential of the present synthetic approach towards the design of inorganic–organic hybrid materials based on PbBr2 and other related metal halide precursors. Moreover, the obtained data show that it is still difficult to control an inorganic fragment which will be preserved (or, to be more exact, will be recreated upon the self-assembly of the coordination compound) from the structure of the inorganic precursor.
Acknowledgements
We are grateful to the University of Maragheh for the financial support of this research. AMK acknowledges the FCT (UID/QUI/00100/2013).Notes and references
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed.
- R. J. Kuppler, D. J. Timmons, Q. R. Fang, J. R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H. C. Zhou, Coord. Chem. Rev., 2009, 253, 3042 CrossRef CAS.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed.
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed.
- X.-P. Li, J.-Y. Zhang, M. Pan, S.-R. Zheng, Y. Liu and C.-Y. Su, Inorg. Chem., 2007, 46, 4617 CrossRef CAS PubMed.
- O. M. Yaghi, H. Li and T. L. Groy, Inorg. Chem., 1997, 36, 4292 CrossRef CAS PubMed.
- C.-L. Chen, B.-S. Kang and C.-Y. Su, Aust. J. Chem., 2006, 59, 3 CrossRef CAS.
- B. D. Wagner, G. J. McManus, B. Moulton and M. J. Zaworotko, Chem. Commun., 2002, 2176 RSC.
- A. M. Beatty, Coord. Chem. Rev., 2003, 246, 131 CrossRef CAS.
- M. Servati-Gargari, G. Mahmoudi, S. R. Batten, V. Stilinović, D. Butler, L. Beauvais, W. S. Kassel, W. G. Dougherty and D. VanDerveer, Cryst. Growth Des., 2015, 15, 1336 CAS.
- L. N. Dawe, T. S. M. Abedin and L. K. Thompson, Dalton Trans., 2008, 1661 RSC.
- L. N. Dawe, K. V. Shuvaev and L. K. Thompson, Inorg. Chem., 2009, 48, 3323 CrossRef CAS PubMed.
- L. N. Dawe, K. V. Shuvaev and L. K. Thompson, Chem. Soc. Rev., 2009, 38, 2334 RSC.
- M. Ruben, J. Rojo, F. J. Romero-Salguero, L. H. Uppadine and J.-M. Lehn, Angew. Chem., Int. Ed., 2004, 43, 3644 CrossRef CAS PubMed.
- M. Ruben, J.-M. Lehn and P. Müller, Chem. Soc. Rev., 2006, 35, 1056 RSC.
- V. A. Milway, S. M. T. Abedin, V. Niel, T. L. Kelly, L. N. Dawe, S. K. Dey, D. W. Thompson, D. O. Miller, M. S. Alam, P. Müller and L. K. Thompson, Dalton Trans., 2006, 2835 RSC.
- G. Mahmoudi, V. Stilinović, M. Servati Gargari, A. Bauzá, G. Zaragoza, W. Kaminsky, V. Lynch, D. Choquesillo-Lazarte, K. Sivakumar, A. Akbar Khandar and A. Frontera, CrystEngComm, 2015, 17, 3493 RSC.
- D. S. Li, Y. P. Wu, P. Zhang, M. Du, J. Zhao, C. P. Li and Y. Y. Wang, Cryst. Growth Des., 2010, 10, 2037 CAS.
- X. L. Wang, Y. Q. Chen, Q. Gao, H. Y. Lin, G. C. Liu, J. X. Zhang and A. X. Tian, Cryst. Growth Des., 2010, 10, 2174 CAS.
- G. P. Yang, L. Hou, Y. Y. Wang, Y. N. Zhang, Q. Z. Shi and S. R. Batten, Cryst. Growth Des., 2011, 11, 936 CAS.
- Z. Y. Du, H. B. Xu, X. L. Li and J. G. Mao, Eur. J. Inorg. Chem., 2007, 4520 CrossRef CAS.
- B. Ding, Y. Y. Liu, X. X. Wu, X. J. Zhao, G. X. Du, E. C. Yang and X. G. Wang, Cryst. Growth Des., 2009, 9, 4176 CAS.
- J. Yang, J. F. Ma, Y. Y. Liu, J. C. Ma and S. R. Batten, Cryst. Growth Des., 2009, 9, 1894 CAS.
- S. H. Li, S. K. Gao, S. X. Liu and Y. N. Guo, Cryst. Growth Des., 2010, 10, 495 CAS.
- T. F. Liu, J. Lu, C. B. Tian, M. N. Cao, Z. J. Lin and R. Cao, Inorg. Chem., 2011, 50, 2264 CrossRef CAS PubMed.
- A. Thirumurugan, R. A. Sanguramath and C. N. R. Rao, Inorg. Chem., 2008, 47, 823 CrossRef CAS PubMed.
- L. Zhang, Z. J. Li, Q. P. Lin, Y. Y. Qin, J. Zhang, P. X. Yin, J. K. Cheng and Y. G. Yao, Inorg. Chem., 2009, 48, 6517 CrossRef CAS PubMed.
- J. D. Lin, S. T. Wu, Z. H. Li and S. W. Du, CrystEngComm, 2010, 12, 4252 RSC.
- E. C. Yang, J. Li, B. Ding, Q. Q. Liang, X. G. Wang and X. J. Zhao, CrystEngComm, 2008, 10, 11 Search PubMed.
- J. Yang, G. D. Li, J. J. Cao, Q. Yue, G. H. Li and J. S. Chen, Chem.–Eur. J., 2007, 13, 3248 CrossRef CAS PubMed.
- K. L. Huang, X. Liu, J. K. Li, Y. W. Ding, X. Chen, M. X. Zhang, X. B. Xu and X. J. Song, Cryst. Growth Des., 2010, 10, 1508 CAS.
- Y. H. Zhao, H. B. Xu, Y. M. Fu, K. Z. Shao, S. Y. Yang, Z. M. Su, X. R. Hao, D. X. Zhu and E. B. Wang, Cryst. Growth Des., 2008, 8, 3566 CAS.
- K. Kavallieratos, J. M. Rosenberg and J. C. Bryan, Inorg. Chem., 2005, 44, 2573 CrossRef CAS PubMed.
- X. R. Meng, Y. L. Song, H. W. Hou, Y. T. Fan, G. Li and Y. Zhu, Inorg. Chem., 2003, 42, 1306 CrossRef CAS PubMed.
- R. L. Davidovich, V. Stavila, D. V. Marinina, E. I. Voita and K. H. Whitmire, Coord. Chem. Rev., 2009, 253, 1316 CrossRef CAS.
- K. L. Zhang, Y. Chang, C. T. Hou, G. W. Diao, R. T. Wu and S. W. Ng, CrystEngComm, 2010, 12, 1194 RSC.
- J. Yang, J. F. Ma, Y. Y. Liu, J. C. Ma and S. R. Batten, Inorg. Chem., 2007, 46, 6542 CrossRef CAS PubMed.
- C. Gabriel, C. P. Raptopoulou, V. Psycharis, A. Terzis, M. Zevou, C. Mateescu and A. Salifoglou, Cryst. Growth Des., 2011, 11, 382 CAS.
- R. L. Davidovicha, V. Stavilab and K. H. Whitmire, Coord. Chem. Rev., 2010, 254, 2193 CrossRef.
- L. Brammer, J. K. Swearingen, E. A. Bruton and P. Sherwood, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4956 CrossRef CAS PubMed.
- M. C. Aragoni, M. Arca, C. Caltagirone, F. A. Devillanova, F. Demartin, A. Garau, F. Isaia and V. Lippolis, CrystEngComm, 2005, 7, 544 RSC.
- G. Ferey, Chem. Mater., 2001, 13, 3084 CrossRef CAS.
- A. K. Cheetham and C. N. R. Rao, Science, 2007, 318, 58 CrossRef CAS PubMed.
- M. Hong, Cryst. Growth Des., 2007, 7, 10 CAS.
- P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638 CrossRef PubMed.
- W. Nieuwenkamp and J. M. Bijvoet, Z. Kristallogr., 1932, 84, 49 Search PubMed.
- A. A. Khandar, V. T. Yilmaz, F. Costantino, S. Gumus, S. A. Hosseini-Yazdia and G. Mahmoudi, Inorg. Chim. Acta, 2013, 394, 36 CrossRef CAS.
- Bruker, SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- Bruker, SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- (a) V. A. Blatov, IUCr Computing Commission Newsletter, 2006, 7, 4 Search PubMed; (b) V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS.
- (a) M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675 CrossRef PubMed; (b) M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- (a) A. Bauzá and A. Frontera, Angew. Chem., Int. Ed., 2015, 54, 7340 CrossRef PubMed; (b) D. Sadhukhan, M. Maiti, G. Pilet, A. Bauzá, A. Frontera and S. Mitra, Eur. J. Inorg. Chem., 2015, 11, 1958 CrossRef; (c) A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317 CrossRef PubMed; (d) P. Chakraborty, S. Purkait, S. Mondal, A. Bauzá, A. Frontera, C. Massera and D. Das, CrystEngComm, 2015, 17, 4680 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- Spartan 10, v. 1.10, Wavefunction Inc, Irvin, CA, USA Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS.
- T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park KS, USA, 2013 Search PubMed.
- The Reticular Chemistry Structure Resource (RCSR) Database; M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 30, 1782 CrossRef PubMed.
- Cambridge Structural Database (CSD, version 2016): F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- M. Servati Gargari, V. Stilinović, A. Bauzá, A. Frontera, P. McArdle, D. Van Derveer, S. W. Ng and G. Mahmoudi, Chem.–Eur. J., 2015, 21, 17951 CrossRef CAS PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314 CrossRef CAS.
Footnote |
| † CCDC 1435154–1435158. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra13462a |
| This journal is © The Royal Society of Chemistry 2016 |