
Theoretical study of the hydrolysis mechanism of dihydrocoumarin catalyzed by serum paraoxonase 1 (PON1): different roles of Glu53 and His115 for catalysis†
Abstract
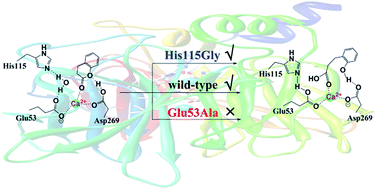
Serum paraoxonase 1 (PON1) is a calcium-dependent enzyme that can catalyze the hydrolysis of multiple substrates, including lactones, thiolactones, carbonates, esters and phosphotriesters, as well as the formation of a variety of lactones. To better understand the lactonase mechanism of PON1, the hydrolysis of dihydrocoumarin, which is considered as a native substrate of PON1, has been investigated by using a combined quantum mechanics and molecular mechanics (QM/MM) approach. Two possible reaction pathways with either Glu53 or His115 acts as the general base have been considered. On the basis of our calculations, these two pathways correspond to the overall energy barriers of 12.5 and 9.0 kcal mol−1, respectively. During the catalytic reaction, if one of the two residues (Glu53 and His115) acts as the catalytic base, the other one forms strong hydrogen bonding interaction with the attacking hydroxide to facilitate the hydrolysis. However, mutation studies reveal that Glu53 is necessary for hydrolysis, whereas His115 is not essential but can promote the activity of PON1. Natural population analysis indicates that the catalytic Ca2+ does not act as Lewis acid but plays structural role in fixing the orientations of the substrate and related residues. In addition, Asp269 is found to coordinate with Ca2+ cation and facilitate the protonation of the alkoxide leaving group by forming hydrogen bond with lactone. These results can explain the fact that mutation of Glu53 results in the loss of activity of PON1, and the hydrolysis of dihydrocoumarin is unaffected by mutation of H115.
 Please wait while we load your content...
Please wait while we load your content...

