
Probing oxygen reduction and oxygen evolution reactions on bifunctional non-precious metal catalysts for metal–air batteries†
Abstract
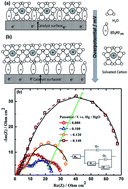
Non-precious metal (NPM) catalysts comprising cobalt and N-doped multiwalled carbon nanotubes (N-MWCNTs-Co) were synthesized by the solid-state pyrolysis (SSP) of melamine with Co3O4. N-MWCNTs-Co acts as an electrocatalyst for both the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER), and hence can be used in secondary metal–air batteries and in unitized regenerative fuel cells. It is important to study the OER and ORR at high concentrations of KOH as most of the metal–air batteries employ KOH concentrations > 4 M. The OER potential of N-MWCNTs-Co at a current density of 10 mA cm−2 was 0.6 V vs. Hg/HgO (corresponding to an overpotential of 0.364 V) in 6 M KOH. The activity towards the ORR of N-MWCNTs-Co at −0.1 V vs. Hg/HgO was 2.7 and 0.2 mA cm−2 in 0.1 M and 6 M KOH respectively. Based on our ORR, OER and impedance studies, we postulate that the non-covalent interactions between the hydrated alkali metal cations and the adsorbed oxygen-based species on the catalyst surface are responsible for the reduction in ORR activity and enhancement in OER activity with increasing KOH concentration. Finally, we provide our initial results using this bifunctional catalyst in a zinc–air battery.
 Please wait while we load your content...
Please wait while we load your content...

