
Preparation of pharmaceutical co-crystals through sustainable processes using supercritical carbon dioxide: a review
Concepción Pando
*,
Albertina Cabañas
and
Isaac A. Cuadra
Dpto. de Química Física I, Facultad C. Químicas, Universidad Complutense, E-28040 Madrid, Spain. E-mail: pando@quim.ucm.es; Fax: +34 91394 4135; Tel: +34 91394 4304
First published on 20th July 2016
Abstract
The preparation of pharmaceutical co-crystals using supercritical CO2 (scCO2) is reviewed. Co-crystallization is an emerging and powerful technique to improve the physicochemical properties of an active pharmaceutical ingredient (API). The solid-state and solution co-crystallization methods usually employed present several disadvantages that may be overcome using the supercritical methods. All the methods employing scCO2 have low environmental impact and operate at relatively low temperature avoiding API degradation and producing solvent-free pharmaceuticals. The role of the fluid varies from one method to another. In the rapid expansion of supercritical solutions (RESS) the API and the coformer are dissolved in scCO2. In the co-crystallization with supercritical solvents (CSS), the fluid also acts as a solvent that facilitates molecular interactions in a suspension of the API and coformer powders. However, in the supercritical antisolvent (SAS) and the gas antisolvent (GAS) crystallizations scCO2 acts as an antisolvent that induces precipitation of the API and the coformer previously dissolved in an organic solvent. In the atomization and antisolvent (AAS) technique and supercritical fluid enhanced atomization (SEA) the fluid may act either as a spray enhancer or an antisolvent depending on the process conditions. Each method is described and its application, advantages and disadvantages are discussed. Future perspectives and areas to be investigated are outlined.
 Concepción Pando | Concepción Pando is Professor of Physical Chemistry at Universidad Complutense de Madrid (UCM), Spain. She received her PhD degree at UCM in 1979 and was a Fulbright scholar at Brigham Young University, USA (1981–82), performing calorimetric studies of mixtures involving supercritical fluids. She has studied phase equilibria and thermodynamic properties of fluid and liquid mixtures formed by CO2. She has experience in supercritical fluid extraction and is currently involved in the fabrication and micronization of materials in supercritical CO2 aimed to applications in catalysis and pharmacology. |
 Albertina Cabañas | Albertina Cabañas is Associate Professor in the Physical Chemistry Department at Universidad Complutense de Madrid (UCM) Spain since 2007. She received her PhD degree at UCM in 1998 performing thermodynamic studies of mixtures involving supercritical fluids. Afterwards, she investigated the synthesis of metal oxide nanoparticles in supercritical water using continuous reactors (1999–2000, University of Nottingham, U.K.) and the deposition of high-quality metal films from supercritical CO2 solutions (2001–2003, University of Massachusets, USA). She was “Ramón y Cajal” Fellow (2003–2007, UCM, Spain). Her current research focuses on the synthesis of nanomaterials using supercritical fluids as green solvents. |
 Isaac A. Cuadra | B.Sc. Degree in Chemical Engineering at the University Complutense of Madrid (UCM), M.Sc. in Industrial Processes Engineering at UCM, Ph.D. student working on the micronization of particles using supercritical carbon dioxide as an antisolvent. Research on enhanced oil extraction at the Research and Development Centre of the Spanish oil company CEPSA. |
1. Introduction
Co-crystallization is an emerging and powerful technique aimed at improving the physicochemical properties of an active pharmaceutical ingredient (API). Properties such as the solubility and chemical stability are modified by establishing a new solid form through the interaction of the API with a coformer at a supramolecular level.1–3 Coformers are usually molecules included on the generally recognized as safe (GRAS) list4 that can interact with the API. Nutraceuticals are another class of compounds with an established safety record that may be used as coformers.5 Pharmaceutical co-crystals resulting from the interaction of two different APIs have been also prepared.6 A co-crystal may be defined as a multiple component crystal that consists of two or more solid components in a definite stoichiometric ratio held together via noncovalent and nonionic interactions. Usually, two components are involved. The co-crystal structure is the result of weak but directional molecular recognition events very often based on hydrogen bonding.7 These intermolecular interactions or synthons are classified either as heterosynthons (e.g. carboxylic acid–amide, pyridine–carboxilic acid synthons) or homosynthons (e.g. carboxylic acid–carboxilic acid, amide–amide synthons). Fig. 1 shows the molecular structures of the anti-inflammatory drug diflunisal and nicotinamide, a member of the vitamin B family widely used as a coformer, together with the supramolecular structure of the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diflunisal–nicotinamide co-crystal showing the heterosynthons involved. This co-crystal has been recently prepared by liquid assisted ball mill grinding and by solution crystallization from ethanol,8,9 and by a method based on using supercritical CO2 (scCO2) as antisolvent.10 The 2:1 diflunisal–nicotinamide co-crystal exhibits an enhanced dissolution rate with respect to that of pure diflunisal.
:1 diflunisal–nicotinamide co-crystal showing the heterosynthons involved. This co-crystal has been recently prepared by liquid assisted ball mill grinding and by solution crystallization from ethanol,8,9 and by a method based on using supercritical CO2 (scCO2) as antisolvent.10 The 2:1 diflunisal–nicotinamide co-crystal exhibits an enhanced dissolution rate with respect to that of pure diflunisal.
 | ||
| Fig. 1 Molecular structures of diflunisal, nicotinamide, and the 2:1 diflunisal–nicotinamide co-crystal showing the supramolecular heterosynthons involved. | ||
Pharmaceutical co-crystals can be prepared by solution methods, evaporative or cooling crystallization or solid-state grinding and mixing.11 Solution co-crystallization is the method preferred if single crystals are desired for crystal structure elucidation although sometimes this method fails in the preparation of a co-crystal already obtained by a mechanochemical method. These preparation methods bear the risk of producing the homocrystals (single component crystalline phases) together with the co-crystal. Either the experimental conditions are established empirically by trial and error or a considerable effort is devoted to obtain ternary phase diagrams of the two components and the solvent that allow understanding the mechanism of a solution co-crystallization.11–13 In addition, remaining residues of the solvents may pose a problem. To overcome some of the difficulties presented by solution and solid state methods, several authors proposed to use co-crystallization methods based on the utilization of supercritical CO2. These supercritical methods are also aimed to the development of production processes with very low environmental impact, fewer steps and a reduced used of organic solvents, therefore fulfilling the requirements for sustainable processes in the pharmaceutical industry. In this paper, the supercritical co-crystallization methods are reviewed, each method is presented in a separate section, and the operating conditions, the properties of the co-crystals obtained so far and the advantages and disadvantages of the methods are examined. Usually, the co-crystals obtained by a supercritical method are compared to those previously prepared by a solid-state grinding or solution method. Another section is devoted to modeling efforts. The state-of art, the future perspectives and the problems related to the application of these methods in the pharmaceutical industries are also discussed. Finally, the possibilities for application of the co-crystallization methods based on supercritical CO2 in other fields different from that of pharmaceutics are summarized.
2. Supercritical carbon dioxide and its use in the field of pharmaceuticals
Supercritical fluids are pure substances or mixtures at a pressure and temperature higher than their critical coordinates, Pc and Tc. Fig. 2 shows the pressure–temperature phase diagram of carbon dioxide and the area where CO2 exists as a supercritical fluid. Carbon dioxide is the most commonly used supercritical fluid because it is nontoxic, non-flammable, inexpensive, has moderate critical temperature and pressure (31.0 °C and 7.39 MPa) and is a byproduct in ethanol and ammonia plants. Therefore, scCO2 is considered a green solvent.14 Supercritical CO2 densities and solvation power are intermediate between those of gases and liquids and can be easily modified with small changes in temperature and pressure, the fluid behavior may be tuned from that of a liquid at a high pressure to that of a gas at low pressure; meanwhile the fluid maintains good transport properties. Values for the scCO2 viscosity coefficient are low and similar to those of gases while values for the diffusivity coefficient are high in comparison to those of liquids. A pressure reduction after scCO2 treatment allows the preparation of solvent-free pharmaceuticals. Other advantages are operation at moderate temperatures in an inert atmosphere thus avoiding the product degradation and the substitution or limited use of organic solvents.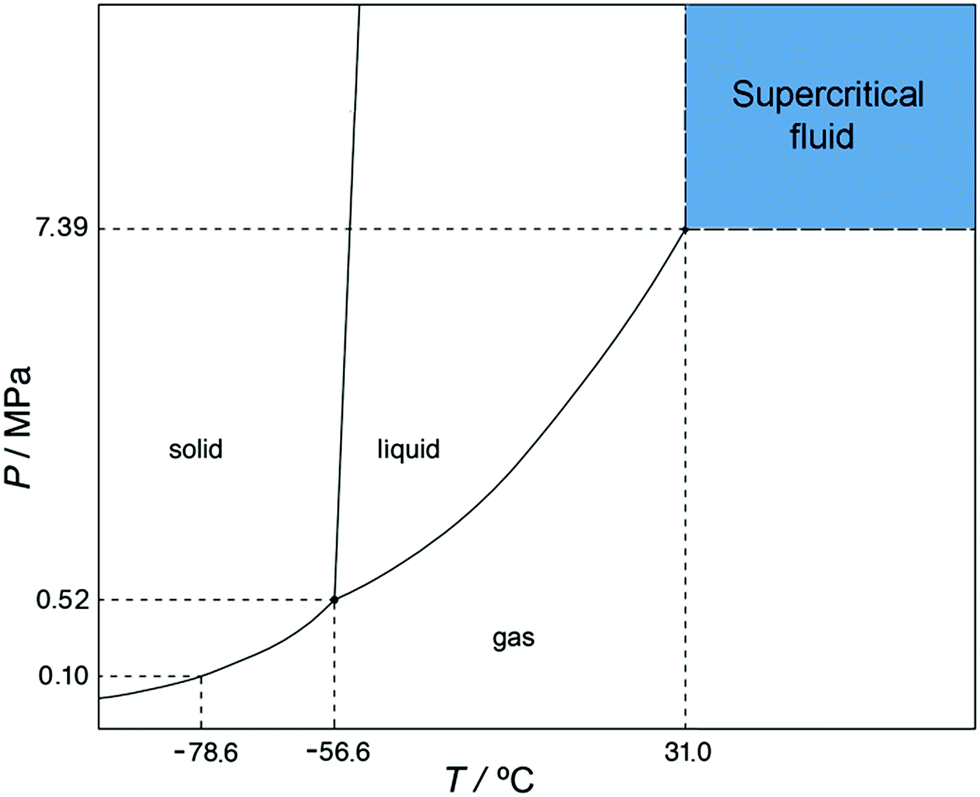 | ||
| Fig. 2 Pressure–temperature phase diagram of carbon dioxide. | ||
Different supercritical techniques have been widely used to improve an API physicochemical properties.15 The solubility and bioavailability of pharmaceuticals may be increased via size reduction. Several supercritical methods for drug micronization are available.16 The possibility of tuning the fluid properties through changes in temperature and pressure allows the control of particle size and morphology. Moreover, the solvent-free particles exhibit narrow size distributions very appropriate for pharmaceutical applications. In comparison with traditional methods, the production of drug microparticles using scCO2 requires fewer steps thus allowing a simplification and better control of the process.
Changes of the crystal habit or polymorphic form through scCO2 treatment are also possible.17–22 APIs can exist as different solid phases classified as polymorphs that differ in crystal structure and exhibit different thermodynamic, spectroscopic and surface properties. Consequently, the various polymorphs of a given API can have different bioavailability, activity and even toxicity. Moreover, the presence of different crystal phases can accelerate undesirable polymorphic transformations. Polymorphic purity is an important parameter in a pharmaceutical product. Conventional crystallization techniques usually lead to a mixture of different polymorphs. However, supercritical fluid techniques may allow the crystallization of a single polymorph because there are additional variables to play with such as the pressure and density of the fluid and furthermore they can be one-step processes.
Very often API formulations consist in a core material (the API) surrounded by a coating or carrier material. In other formulations the API particles are embedded in a carrier material. Usually, a natural or synthetic polymer is used to prepare both types of formulations, although fats or sugars may be also used.23 Composite microparticles present several advantages, in the first place, they are easier to handle and dose than the pure particles of an API. The material selected as shell/carrier depends on the intended use of the composite microparticles and improves its properties. A biodegradable carrier is used to achieve a controlled delivery of the active ingredient into the targeted media.24,25 If the API is susceptible of degradation, the carrier acts as a protection against aggressive agents. If the API is poorly soluble in the targeted media, the carrier facilitates its solubilization. This is the case of a number of poorly water-soluble drugs. For drugs with low solubility in the crystalline state, the possibility of improving their solubility, dissolution rate and bioavailability due to the formation of an amorphous form in a solid dispersion presents additional advantages. On the other hand, in some cases composite particles exhibit smaller sizes than that of the pure drug processed under the same conditions.26 These formulations can be prepared by coprecipitation, encapsulation or impregnation in scCO2 or extraction of an emulsion using scCO2.15,23–31
The adaptation of the already existing supercritical techniques and the proposal of new ones aimed to the preparation of pharmaceutical co-crystals started in 2002;32 its fast development in the last five years10,32–51 prompted us to undertake this review. The supercritical co-crystallization methods are listed in Table 1; all of them share the advantages associated to the use of CO2 already mentioned and have specific advantages of their own that will be discussed and compared. The order of presentation of the methods follows the chronological order of the application of each method in the literature. The role of scCO2 differs from one method to another. In the rapid expansion of supercritical solutions (RESS) the API and the coformer are dissolved in scCO2. In the co-crystallization with supercritical solvent (CSS), the fluid also acts as a solvent that facilitates molecular interactions and thereby nucleation and growth of co-crystals in a suspension of the API and coformer powders. However, scCO2 acts as an antisolvent that induces precipitation of the API and the coformer previously dissolved in an organic solvent in the supercritical antisolvent (SAS) and gas antisolvent (GAS) crystallizations. In the atomization and antisolvent (AAS) and in the supercritical fluid enhanced atomization (SEA) techniques the fluid has the dual role of antisolvent and spray enhancer. Prior to the description of the supercritical methods and in order to present the results of their application, it is necessary to briefly review the techniques involved in the evaluation of pharmaceutical co-crystals.
| Method | Acronym | Role of CO2 | Reference |
|---|---|---|---|
| a Two co-crystallization patents should be added to this table: Mazen and Townend50 filed a general process including GAS, SAS and SEDS (solution enhanced dispersion by supercritical fluids); Hofland and Van Rosmalen51 patented a method similar to CSS. | |||
| Rapid expansion of supercritical solutions | RESS | Solvent | Vemavarapu et al. 2002,32 Vemavarapu et al. 2009,33 Müllers et al. 2015 (ref. 46) |
| Co-crystallization with supercritical solvent | CSS | Solvent and molecular mobility enhancer | Padrela et al. 2009,34 Padrela et al. 2015 (ref. 44) |
| Supercritical antisolvent crystallization | SAS | Antisolvent | Padrela et al. 2009,34 Chen et al. 2015,47 Cuadra et al. 2016,10 Hiendrawan et al. 2016,48 Neurohr et al. 2016 (ref. 49) |
| Atomization and antisolvent crystallization | AAS | Spray enhancer or antisolvent | Padrela et al. 2009 (ref. 34) |
| Supercritical fluid enhanced atomization | SEA | Spray enhancer or antisolvent | Padrela et al. 2010,35 Tiago et al. 2013,40 Padrela et al. 2014 (ref. 41) |
| Gas antisolvent crystallization | GAS | Antisolvent | Shikhar et al. 2011,36 Ober and Gupta 2012,37 Ober et al. 2013,38 Neurohr et al. 2013,39 Harscoat-Schiavo et al. 2015,42 Neurohr et al. 2015,43 Erriguible et al. 2015 (ref. 45) |
3. Evaluation of pharmaceuticals co-crystals
The methods used to evaluate pharmaceuticals co-crystals prepared using scCO2 do not differ from those used for co-crystals prepared by conventional methods. Most co-crystals obtained using scCO2 have been previously obtained by these methods and the co-crystal characterization available in the literature serves as a guide to evaluate the co-crystal obtained by the supercritical method. The characterization methods are aimed to study the crystal size and morphology, its purity, structure and interactions and the physical properties such as the dissolution rate involved in the co-crystal pharmacological use.1 Crystal size and morphology are usually investigated by scanning electron microscopy (SEM). Optical microscopy has been also used. Particle size distribution may be obtained from SEM images or in a separate laser diffraction experiment. The co-crystal size is an important characteristic although micronization is not the principal objective in the scCO2 co-crystallization techniques. The pharmacological benefits are expected to derive from the new formulation. Nevertheless, a small particle size would also help in the case of poorly-soluble APIs.The composition of the powders produced is a key issue. The coformers homocrystals may precipitate together with the desired co-crystal. High performance liquid chromatography (HPLC) may be used to establish the co-crystal purity. Using the adequate mass balance equations, the amount of co-crystals in the produced power can be determined. On the other hand, when organic solvents are required the absence of solvent residues should be confirmed. Differential scanning calorimetry (DSC), infrared spectroscopy (FTIR) and X-ray diffraction (XRD) are used to establish the co-crystal structure and interactions. X-ray diffraction (XRD) of single crystals obtained from solution co-crystallization is the preferred tool to determine the solid state structure of a given co-crystal. Powder or microcrystalline samples are usually the result of supercritical methods. Therefore, if a new co-crystal is obtained by a supercritical method a parallel solution preparation is recommended. Afterwards, XRD is used to identify the co-crystal and to study the commercial coformers and establish their polymorphic form. Padrela et al. also proposed a method for the quantification of co-crystals in the precipitate based on power XRD data.52 DSC is used to obtain melting point data and thermal data such as the enthalpy of melting. Usually, the co-crystal melting point is either comprised between those of the API and the coformer or lower than both of them.1 DSC is also used to assess the co-crystal purity. The appearance of one or two peaks corresponding to the homocrystals together with the co-crystal peak indicates that conditions of the co-crystallization method need to be modified. Raman spectroscopy is sometimes used to complement the DSC and XRD findings. Interactions between the API and the coformer in the co-crystal are investigated using FTIR. The functional groups in the co-crystal spectrum show differences in the wavenumbers and intensity of the vibrational modes in comparison to those of the two coformers individual spectra. Furthermore, bands characteristic of the synthons established are observed. For instance, for the diflunisal–nicotinamide co-crystals whose structure is shown in Fig. 1 bands characteristic of the acid–pyridine nitrogen synthon and the carboxylic acid–amide hydrogen bonds were observed in the co-crystal spectrum.10
The determination of the solubility of APIs and coformers in scCO2 may be necessary in order to optimize the experiments when solubility data are not available in the literature. Also, phase diagrams for the quaternary systems formed by the API, the coformer, a solvent and CO2 are required in order to understand the co-crystallization mechanism. This type of data is not directly related to the crystal evaluation although it is essential to understand the co-crystallization mechanisms.
Co-crystal dissolution profiles need to be obtained and compared to those of the API. For poorly soluble APIs an improvement in the dissolution rate is considered a benefit of the new formulation. Physical and chemical stability studies are also conducted; relative humidity stability experiments need to be carried out if hygroscopicity leads to form changes or degradation. Finally, the yield should be also studied because it gives an idea whether the process can be applied in the industry.
4. Rapid expansion of supercritical solutions, RESS
In the RESS process solutes are dissolved in supercritical CO2 forming a homogeneous supercritical solution. Solute nucleation is induced by rapidly reducing the solution density through expansion to atmospheric conditions. This expansion causes a high supersaturation that leads to a very homogeneous nucleation rate. Fig. 3 shows a scheme of a RESS setup.32 Liquid CO2 is pressurized using a pump and is fed to a preheater where the temperature of the pressurized liquid is raised to that of a supercritical fluid. scCO2 is then fed to the saturation vessel that contains the solutes to be co-crystallized and glass beads aimed at improving the extraction efficiency and buffering the turbulent flow of the fluid through the vessel. The saturated fluid solution flows from the temperature-controlled reaction vessel into a pre-expansion chamber maintained at 50 degrees above the extraction temperature. Temperature is also controlled in the lines and connectors to avoid premature precipitation of the solutes. The saturated solution passes through a micrometering valve maintained at 100 to 150 °C prior to the rapid expansion. The expansion device is a stainless steel capillary inserted through the cap of the collection vial. After solid precipitation CO2 is exhausted through a filter and passes through a 5 m tubing before entering a flow meter that allows to obtain a reliable estimate of the average flow rates through the system.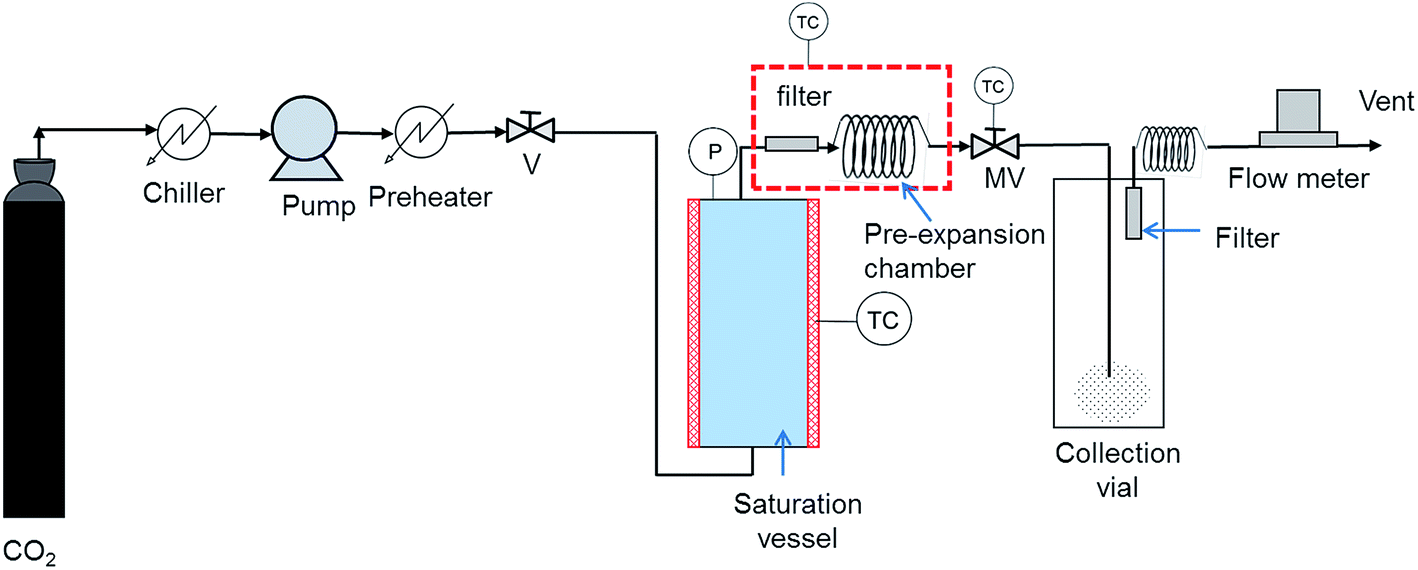 | ||
| Fig. 3 Schematic representation of a RESS process:32 (V) valve, (P) pressure measurement, (TC) temperature controller, (MV) micrometering valve. | ||
Although many APIs are scarcely soluble in scCO2 the RESS method has been used to produce micro and nanoparticles of several pharmaceuticals.16 As to co-crystals, the rapid expansion of chlorpropamide–urea–CO2 solutions was used by Vemavarapu et al. in 2002 (ref. 32) to disrupt the crystalline structure of chlorpropamide with urea. Chlorpropamide is an API poorly soluble in water that belongs to the sulfonylurea hypoglycemic agents. Although the aim of Vemavarapu et al. was crystal doping, this study can be considered the first attempt to obtain a co-crystal using scCO2. RESS recrystallization of commercial chlorpropamide (polymorph A) resulted in polymorph conversion to forms C and V depending on the temperature and pressure. Temperature was varied from 46 to 103 °C and two levels of pressure (27.6 and 55.2 MPa) were tried at each temperature. The evidence of crystal disruption through RESS was confirmed by changes in the XRD patterns and the lowering of the melting point in comparison to those of the pure crystal polymorphs. SEM images revealed a particle size reduction of up to one order of magnitude as a consequence of RESS processing. The doped crystals thus formed are expected to show a dissolution rate enhancement. In contrast, chlorpropamide recrystallizations from liquid organic solvents lacked the ability to produce polymorphic changes. Also, the incorporation of urea into the drug lattice was found to be insufficient. Although minor reductions in the melting temperatures and the heat of fusion values were observed with respect to those of pure chlorpropamide, powder XRD patterns revealed no significant disruption in the crystallinity. A few years later, Vemavarapu et al.33 used this technique for the coprecipitation of twelve drug-additive mixtures and observed several phenomena including co-crystal and hydrate formation and polymorphic transitions. Temperature was varied from 35 to 100 °C and pressure was varied from 7.6 and 62.1 MPa. According to Vemavarapu et al., different crystallization mechanisms competed depending on factors such as the interactions between the drug and the coformer and their relative solubility in the supercritical fluid. In this study, the composition of several co-crystals of theophylline and caffeine was determined by HPLC and found to be dependent on that of the starting mixture. Depending on the relative amount of API and additive in the co-crystal varying levels of crystallinity ranging from pure crystals to amorphous mixtures could be found. These results led these authors to conclude that RESS could not be used to consistently dope crystals.
Recently, Müllers et al.46 have used RESS to obtain co-crystals with coformers that have sufficient solubility in scCO2. Ibuprofen and nicotinamide were dissolved in scCO2 at 30 MPa and 50 °C with molar ratios of 0.5:1, 1:1 and 2:1. The difference in solubility was taken into account in order to obtain a supercritical solution with the correct stoichiometry. The co-crystals obtained were thoroughly characterized. A 1:1 ibuprofen–nicotinamide co-crystal of high purity was produced with a yield of 20%. The RESS co-crystal was shown to be equivalent to the co-crystal prepared by slow solvent evaporation. The crystalline micronized co-crystal product was produced by RESS in a one-step process without the need of organic solvents or further processing such as drying or size reduction. The disadvantages of the RESS method are the low solubility of many APIs in scCO2 that often requires the use of high pressures and the low yield and throughput rates. A mole fraction solubility of at least 10−4 is required for RESS crystallization of a pure API.16 As to the low throughput rates, Müllers et al. suggested that they could be improved by implementing a continuously running process.
5. Supercritical antisolvent crystallization, SAS
The SAS method is based on the relatively low solvent power of scCO2 for solutes such pharmaceuticals and its good miscibility with many organic solvents. The API and the coformer are simultaneously dissolved in a polar organic solvent such as ethanol or acetone and scCO2 is used as antisolvent to induce the co-crystal precipitation. SAS has been widely applied to obtain pharmaceutical particles.16 As to co-crystals, the group of Prof. Gomes de Azevedo was the first to apply this method in the preparation of indomethacin–saccharin co-crystals.34 Recently, Cuadra et al.,10 Chen et al.,47 Hiendrawan et al.,48 and Neurohr et al.49 prepared by this method piracetam–salicylic acid, diflunisal–nicotinamide, paracetamol–dipicolinic acid, and naproxen–nicotinamide co-crystals, respectively.A scheme of the SAS setup is shown in Fig. 4.10 Supercritical carbon dioxide is introduced in a precipitation chamber using a high-pressure pump at constant flow rate. Then the organic solution containing the drugs is fed through a second pump also at a constant flow rate reaching steady state operating conditions and an adequate supercritical fluid/solvent ratio. A given amount is sprayed in the precipitation chamber through a nozzle. The chamber is heated and both temperature and pressure are controlled. When the fluid dissolves in the solution, the mixture becomes supersaturated and precipitation starts. The co-crystals are collected at the bottom and walls of the precipitation chamber. At the end of the precipitation process, the chamber is washed with excess CO2 to remove the residual content of the liquid solvent. The CO2 + organic solvent mixture is introduced into a separation chamber where the solvent is recovered. The solvent-free particles exhibit narrow size distributions. The pharmaceutical co-crystals obtained using SAS are listed in Table 2 together with the solvent, temperature, pressure, concentration, and the mass flow ratio of the solution to the supercritical fluid used.
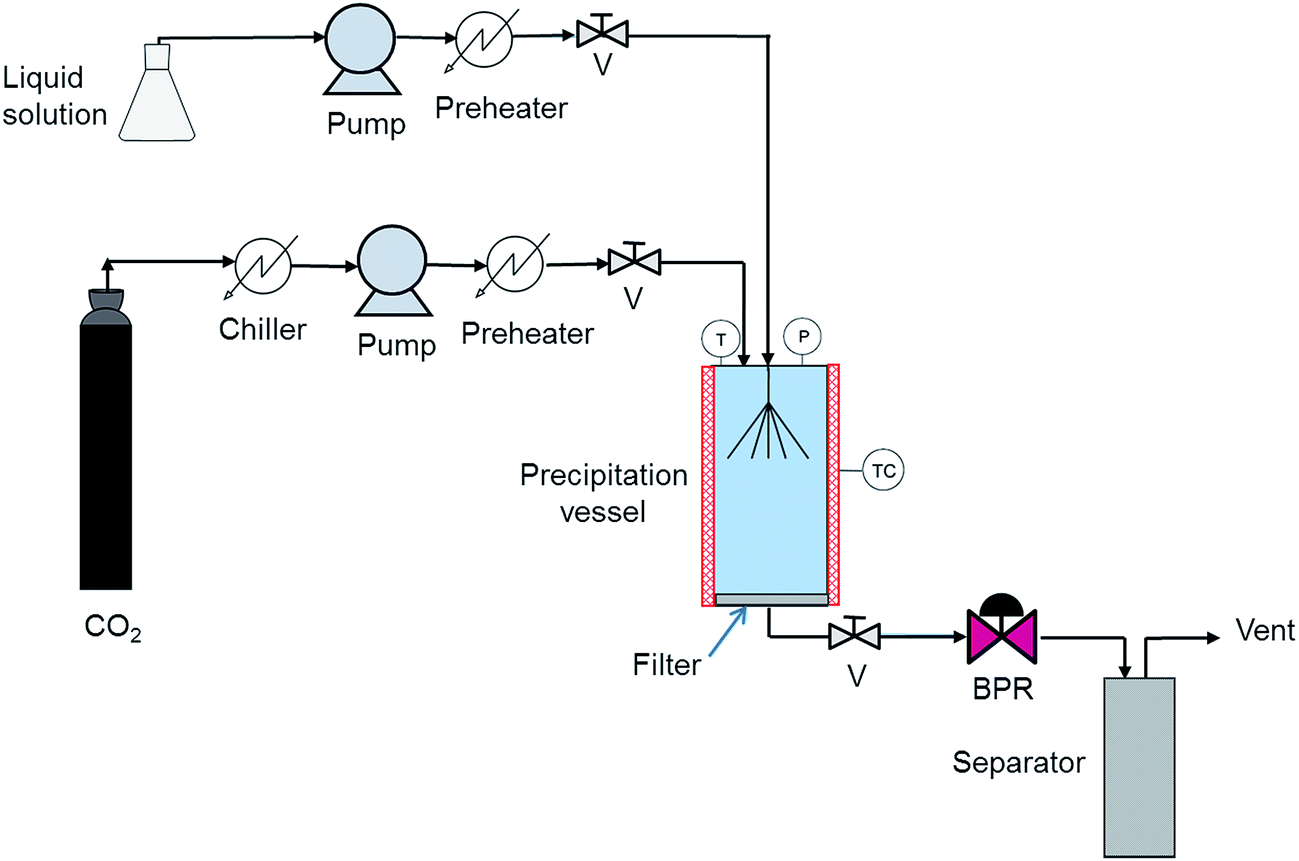 | ||
| Fig. 4 Schematic representation of a SAS setup:10 (BPR) back pressure regulator, (P) pressure measurement, (V) valve, (TC) temperature controller, (T) temperature measurement. | ||
| Co-crystal | Solvent | T (°C) | P (MPa) | R (g g−1) | Reference |
|---|---|---|---|---|---|
| a In the case of the naproxen–nicotinamide co-crystal a 2:2 molar ratio was also used.b Two different concentration values were used. |
|||||
| 1:1 indomethacin–saccharin |
Ethanol | 50 | 9.0 | 0.05 | 34 |
| Acetone | 50 | 8.4 | 0.12 | 34 | |
| Tetrahydrofurane | 50 | 8.5 | 0.04 | 34 | |
| Methanol | 50 | 8.6 | 0.06 | 34 | |
| Ethyl acetate | 50 | 8.6 | 0.19 | 34 | |
| 2:1 diflunisal–nicotinamide |
Acetoneb | 35 | 10.0 | 0.04 | 10 |
| Acetoneb | 35 | 12.0 | 0.04 | 10 | |
| Acetoneb | 40 | 10.0 | 0.04 | 10 | |
| Acetoneb | 40 | 12.0 | 0.04 | 10 | |
| Ethanol | 35 | 10.0 | 0.04 | 10 | |
| 1:1 piracetam–salicylic acid |
Acetone | 45 | 10.0 | — | 47 |
| 1:1 paracetamol–dipicolinic acid |
Methanol | 40 | 10.0 | 0.03 | 48 |
| 2:1 naproxen–nicotinamideb |
Acetone | 37 | 10.0 | 0.09–0.50 | 49 |
Cuadra et al.10 used the SAS setup shown in Fig. 4 to obtain co-crystals of the anti-inflammatory drug diflunisal and nicotinamide. The drug and coformer molar ratio in solutions followed the 2:1 stoichiometric co-crystal composition. The influence of solvent (acetone and ethanol), concentration (two levels), temperature (35 and 40 °C) and pressure (10.0 and 12.0 MPa) was shown to be weak. Fig. 5 shows the SEM image of the co-crystal obtained at 35 °C, 10.0 MPa, and concentrations in the acetone solution of 30.00 and 7.28 mg mL−1 for diflunisal and nicotinamide, respectively. A similar crystalline material in the form of needles of uniform width was obtained in all experiments. A yield of 70% was obtained for the co-crystal prepared with the lower values of concentration in acetone at the lower values of temperature and pressure. This pure co-crystal was shown to exhibit the same crystal structure, melting point and FTIR spectrum as co-crystals previously obtained by assisted ball mill grinding and solution crystallization. The dissolution profile of the 2:1 diflunisal–nicotinamide co-crystal was better than that of pure diflunisal. The application of the SAS technique to pure diflunisal led to a change in the diflunisal polymorphic form.
 | ||
| Fig. 5 SEM image of the 2:1 diflunisal–nicotinamide co-crystal obtained by SAS at 35 °C, 10.0 MPa, and concentrations in the acetone solution of 30.00 and 7.28 mg mL−1 for diflunisal and nicotinamide, respectively (2:1 molar ratio). | ||
Chen et al.47 obtained the 1:1 piracetam–salycilic acid co-crystal at the conditions shown in Table 2. The co-crystal was characterized by SEM, XRD, FTIR, DSC and thermogravimetric analysis (TGA). The formation of the co-crystal and its satisfactory thermal stability were confirmed. Dissolution studies indicated that at a given time the co-crystal concentration is close to half of that for pure piracetam. Therefore, the side effect caused by a high dissolution rate of pure piracetam can be regulated.
Hiendrawan et al.48 obtained the 1:1 paracetamol–dipicolinic acid co-crystal at the conditions shown in Table 2 and compare it to that obtained by a traditional solvent evaporation method. The formation of new crystalline phases was confirmed by XRD, DSC, FTIR, SEM and polarized light microscopy. The SAS co-crystal particles had a mean diameter of 4.18 μm (considerably smaller than that of solvent-evaporation particles) and showed an enhanced dissolution rate with respect to both pure paracetamol and the co-crystal produced by solvent evaporation. Both co-crystals were found to be stable and exhibited better tableting properties than pure paracetamol.
Using a SAS setup similar to that shown in Fig. 4, Neurohr et al.49 have recently prepared the 2:1 naproxen–nicotinamide co-crystals. An acetone solution and CO2 were introduced into a cylindrical vessel through two separated inlet ports. The vessel was equipped with 8 sapphire windows distributed at top, medium and bottom levels to visualize the injection and the precipitation. The conditions of temperature and pressure were kept constant at 37 °C and 10 MPa while the CO2 and solution flow rates were varied. The co-crystal particles exhibited a thin plate-like morphology, a size distribution ranging from 20 μm to 1 mm. Also, the SAS co-crystals showed the same hydrogen bond interactions and crystalline structure than co-crystals previously obtained by conventional and GAS techniques.42,43,45 By processing a 2:2 naproxen–nicotinamide solution instead of a 2:1, Neurohr et al. obtained a co-crystal-pure powder. The yield of precipitation ranged from 60 to 70%. A simulation was developed to obtain supersaturation distribution profiles that helped to establish conditions favoring co-crystallization instead of homo-crystallization. The solubility data previously obtained by Revelli et al.53 for the quaternary system naproxen–nicotinamide–acetone–CO2 were taken into account. More details about this simulation are given in Section 10.
Fig. 6 shows a scheme of the SAS setup used in the experiments carried out by Gomes de Azevedo and coworkers34 which has the versatility to be used for three different micronization techniques using CO2: the SAS process and the AAS and SEA processes which will be described in the next sections. The supercritical fluid is introduced in the precipitation vessel until the desired pressure is reached, being regulated by a metering valve located at the exit. Next, the solution containing indomethacin and saccharin is pumped through a coaxial nozzle that includes three sections: the inlet where the two lines for each phase are introduced, a small mixing chamber where solution and fluid mix prior to their full mixing inside the precipitation vessel and the orifice disk where droplets are formed. The pre-expansion pressure (before de nozzle) is approximately 1 MPa higher than the post-expansion pressure (after the nozzle) that is listed in Table 2. Indomethacin–saccharin co-crystals were obtained at 50 °C using 1:1 molar ratio solutions in five different solvents, at pressure values of approximately 9 MPa and mass flow ratio of the solution to the supercritical fluid ranging from 0.04 to 0.19 g g−1. DSC, XRD and Raman spectroscopy were used to identify/characterize and estimate the purity of the co-crystalline phase that was shown to be the same as that one previously obtained by a solution method. SEM analysis revealed a mixture of needle and block shaped microparticles. Padrela et al. attributed these two morphologies to competing mechanisms in SAS co-crystallization. No remarkable differences were observed in the microparticles obtained at different processing conditions.
 | ||
| Fig. 6 Schematic representation of a AAS, SAS and SEA apparatus:34 (MV) metering valve, (TC) temperature controller, (P) pressure measurement, (T) temperature measurement. | ||
The one-step SAS process has the advantage of being applicable for APIs and coformers poorly soluble in scCO2. The use of a polar solvent may be mentioned as a disadvantage. Nevertheless, this solvent is easily separated and solvent-free co-crystals with a narrow size distribution are obtained. Yield values similar to that of 70% reported by Cuadra et al.10 and Neurohr et al.49 and to those found for other pharmaceutical crystals obtained by SAS may be expected.
6. Co-crystallization with supercritical solvent, CSS
Padrela et al. observed in 2009 that indomethacin–saccharin co-crystals could be obtained if powders of the two coformers in a 1:1 molar ratio were suspended in scCO2.34 Pure co-crystals could not be prepared and Padrela et al. attributed this fact to the poor dissolution of the coformers in scCO2. Recently, Padrela et al.44 further explored this co-crystallization technique and found conditions for preparing pure co-crystals. A scheme of the CSS setup is shown in Fig. 7. The pure co-crystals obtained via the CSS process and the experimental conditions used are listed in Table 3. The desired amounts of the two powders are placed in a stainless steel high-pressure vessel with monitored temperature and pressure. Since suspensions replace the polar solvent solutions used in GAS or SAS, the size of this high-pressure vessel is relatively small. Prior to its introduction into it, the CO2 is loaded using a compressor into a second vessel that acts as a CO2 reservoir. Both vessels are placed inside a temperature-controlled air chamber. In a typical experiment the API and the coformer are mixed with CO2 at 20.0 MPa and 50 °C under magnetic stirring for two hours. Then the high-pressure vessel is slowly depressurized and the resulting material is collected. In the CSS method, the fluid acts as a solvent that facilitates molecular interactions and thereby nucleation and growth of co-crystals in despite of the low solubility of the coformers in scCO2. Nevertheless, similarly to the RESS method, the CSS method relies on the solvent power of scCO2 and its application requires that the API and the coformer have sufficient solubilities in scCO2.
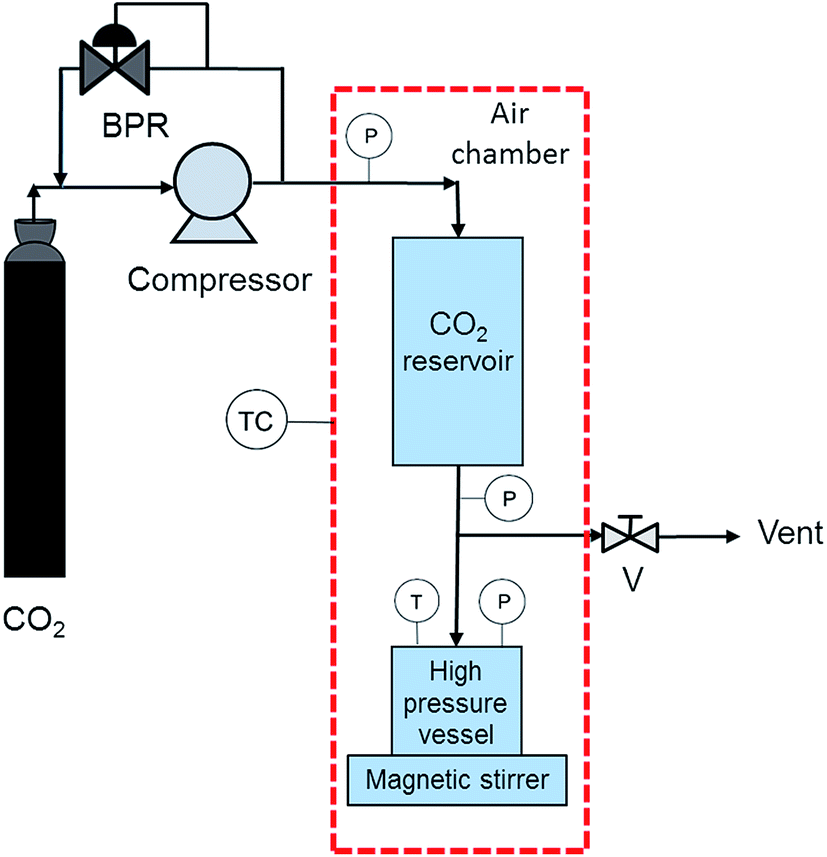 | ||
| Fig. 7 Schematic representation of a CSS setup:44 (BPR) back pressure regulator, (P) pressure measurement, (V) valve, (TC) temperature controller, (T) temperature measurement. | ||
| Co-crystal | Suspension molar ratio | T (°C) | P (MPa) | Cosolventa | Reaction time (h) | Reference |
|---|---|---|---|---|---|---|
| a 1 μL of ethanol per mg of powder was added.b Reaction time was varied from 0 to 2 h. The 2 h experiment was repeated without stirring.c The experiment was repeated without stirring. | ||||||
| 1:1 indomethacin–saccharin |
1:1 |
50 | 14 | — | 3.5–90 | 34 |
| 1:1 |
50 | 22 | — | 22 | 34 | |
| 0.9:1 |
50 | 14 | — | 20 | 34 | |
| 1:1 |
50 | 20.0 | — | 0–2.0b | 44 | |
| 1:1 |
50 | 20.0 | Ethanol | 2.0 | 44 | |
| 1:1 theophylline–saccharin |
1:1 |
50 | 20.0 | — | 0–2.0b | 44 |
| 1:1 |
30 | 20.0 | — | 2.0 | 44 | |
| 1:1 |
40 | 20.0 | — | 2.0 | 44 | |
| 1:1 |
50 | 8.0 | — | 0–2.0 | 44 | |
| 1:1 |
30 | 8.0 | — | 2.0 | 44 | |
| 1:1 |
40 | 8.0 | — | 2.0 | 44 | |
| 1:1 |
30 | 8.0 | — | 0–2.0 | 44 | |
| 1:1 |
50 | 20.0 | Ethanol | 2.0 | 44 | |
| 1:1 carbamazepine–saccharin |
1:1 |
50 | 20.0 | — | 2.0c | 44 |
| 1:1 |
50 | 20.0 | Ethanol | 2.0 | 44 | |
| 1:1 caffeine–saccharin |
1:1 |
50 | 20.0 | — | 2.0c | 44 |
| 1:1 |
50 | 20.0 | Ethanol | 2.0 | 44 | |
| 1:1 sulfamethazine–saccharin |
1:1 |
50 | 20.0 | — | 2.0c | 44 |
| 1:1 |
50 | 20.0 | Ethanol | 2.0 | 44 | |
| 1:1 acetylsalicylic acid–saccharin |
1:1 |
50 | 20.0 | — | 2.0c | 44 |
| 1:1 |
50 | 20.0 | Ethanol | 2.0 | 44 | |
Padrela et al.44 have studied the mechanism of the CSS process using the theophylline–saccharin as a model system. Convection was revealed as the key parameter for successful co-crystallization with high yield. Different conditions of pressure (8–20 MPa), temperature (30 to 50 °C) and convection regimes were selected to study the CSS kinetics in the gas, supercritical and liquid phase. The effect of temperature and pressure on the co-crystallization yield after 2 h in CO2 was investigated. High values were obtained at 50 °C and pressures higher than the CO2 critical pressure. On the other hand, experiments carried out at constant pressures of 8.0 and 20.0 MPa and temperatures increasing from 30 to 50 °C revealed a slight decrease in the yield as temperatures rise which may attributed to the decrease in the CO2 density. Co-crystallization rates improved as the CO2 density increased. When different APIs were studied with saccharin as coformer (namely, indomethacin, carbamazepine, caffeine, sulfamethazine and acetylsalicylic acid), the rate improved for the API more soluble in CO2. Therefore, a dissolution step seems to be involved in the co-crystallization process. The use of CO2 together with the cosolvent ethanol allowed the preparation of co-crystals that could not be obtained in pure CO2. Some co-crystals could be also obtained using supercritical N2 instead of CO2. However, the latter was shown to be a better media for the co-crystallization process. Equimolar amounts of the API and the coformer saccharin, and a 300 rpm stirring were used in all cases. Homogeneity in the co-crystallization medium caused by stirring was determinant for a successful co-crystallization due to the enhancement of molecular interactions between the two coformers. When stirring was omitted either a mixture of the API and saccharin or a mixture of co-crystal, API and saccharin was obtained.
CSS is a novel, simple and solvent-free batch process that presents the additional advantage of using a small reactor thus minimizing the volume under pressure, which is a problematic factor for scale up. In 2009 Hofland and Van Rosmalen51 patented a method similar to the CSS method in which the API and the coformer are brought simultaneously into contact with a supercritical or liquefied gas.
7. Atomization and antisolvent crystallization, AAS
The AAS technique proposed by Padrela et al.34 is closely related to the SAS and SEA techniques and the same apparatus shown in Fig. 6 is used to carry out the three processes. The solution containing the solutes is pumped through a coaxial nozzle into the mixing chamber where it mixes with scCO2 prior to its depressurization in a precipitation vessel. In the SAS technique the precipitator is filled with CO2 at high pressure while in the AAS technique it is at ambient pressure. CO2 may be replaced by N2. Also, a secondary stream of dry N2 is fed into the precipitation vessel to enhance the solvent extraction from the particles. In order to prepare indomethacin–saccharin co-crystals, a 1:1 molar ratio mixture of indomethacin and saccharin was dissolved in different solvents: ethanol, acetone, tetrahydrofurane, methanol and ethyl acetate. The pre-expansion pressure, before de nozzle, was varied from 6 to 12 MPa and the post-expansion pressure was comprised between 0.1 and 0.9 MPa. The mixing chamber temperature was set between 50 and 70 °C. The mass flow-rate ratio of the solution and the scCO2 was varied from 0.03 to 0.19 g g−1. DSC, XRD, and Raman spectroscopy were used to identify/characterize and estimate the purity of the co-crystalline phase that was shown to be the same as that one previously obtained by a solution method. Pure indomethacin–saccharin co-crystals were prepared by AAS. No remarkable differences were observed in the microparticles obtained at different processing conditions. SEM images showed small spherical co-crystal particles with similar size distributions obtained using a laser diffraction aerosizer. Average diameter values ranged from 0.5 to 0.7 μm. Padrela et al. attributed this uniform behavior to similarities in the solvents used and a common co-crystallization mechanism. In a previous study, Rodrigues et al.54 showed that, depending on the pre-nozzle conditions in the AAS process with CO2, crystallization may be governed by supercritical antisolvent or spray drying. In the indomethacin–saccharin co-crystal experiments the spray drying mechanism prevails.
8. Supercritical fluid enhanced atomization, SEA
The supercritical fluid enhanced atomization proposed by the group of Prof. Gomes de Azevedo35,40,41 is based on the supercritical fluid ability to enhance the breakup of liquid jets into fine droplets when depressurized simultaneously with liquid solutions. SEA is closely related to the solution enhanced dispersion by supercritical fluids (SEDS) or the supercritical assisted atomization (SAA) that have been used previously to produce pharmaceutical microparticles16 and the AAS technique described in the previous section. The SEA setup is essentially that shown in Fig. 6 for SAS and AAS. CO2 is compressed until the desired pressure using a gas compressor and is kept in a temperature-controlled gas storage cylinder. The coaxial nozzle is essential for SEA and its flow is measured by a mass flow meter. Temperatures are controlled inside the air chamber or in a water bath. A tetrahydrofuran or ethanol solution containing the API and the coformer is pumped through the nozzle where it mixes with scCO2 before depressurization in a precipitation vessel. A considerable volume reduction (10 times) takes place causing a strong pre-nozzle acceleration that impulses the liquid breakup. Therefore, the purpose of the small mixing chamber is the atomization enhancement due to the high density and velocity of the supercritical fluid phase. It is not necessary to saturate the solution with the antisolvent; this process does not rely on the miscibility of both phases. The temperature of the precipitation vessel (usually 50 °C although values of 40, 60 and 70 °C were also used) is controlled and droplets are dried at this temperature near atmospheric pressure (0.1–0.5 MPa). The liquid/gas mass flow ratio was chosen to be below 50% solvent saturation at atmospheric CO2 at 50 °C. Particles are collected in the precipitator walls and from a filter at the precipitator exit. Critical data for the CO2 + ethanol or tetrahydrofuran mixtures are required to control the coprecipitation mechanism by choosing a value for the pressure of the mixture before the nozzle. Above the critical pressure particles may precipitate by the CO2 antisolvent effect, while below the critical pressure supersaturation is driven by spray drying.54 The co-crystals obtained by SEA and the experimental conditions used are listed in Table 4. In contrast to AAS, SEA does not require the use of an additional stream of N2 to enhance the solvent extraction from particles. In a first study Padrela et al.35 obtained co-crystals of six different APIs with saccharin using ethanol as solvent, a temperature of 50 °C and a pressure before the nozzle of approximately 8 MPa. Co-crystals were characterized by DSC, XRD, laser diffraction aerosizer and SEM. A new co-crystalline form was found for theophylline–saccharin. No residual peaks of homocrystals were observed in the DSC thermograms. Particle size distributions were in the 0.3–10 μm range. The 1:1 theophylline–saccharin co-crystal was later obtained at 50 °C and 8.0 MPa using tetrahydrofurane as solvent.40 This co-crystal was shown to extend the API stability at 92% relative humidity by 6 months with respect to that of the pure API. Dispersions of theophylline and the co-crystal in hydrogenated palm oil were also prepared. Padrela et al.41 also prepared and characterized the co-crystals of theophylline and different coformers using tetrahydrofurane as solvent. Particle size distributions were in the 0.3–5 μm range. The influence on the size distributions of the SEA process variables, the chosen coformer, the concentration of API and coformer, the value of pressure before the nozzle, and the temperature in the mixing chamber was studied and the temperature was shown to be the only variable with a significant influence. Mean particle sizes increased as temperature rises. The nature of the coformer did influence the particles morphology and dissolution rate. The solubility of each coformer in a phosphate buffered medium is related to the dissolving rate behavior of the co-crystal. Coformers with low solubility generate co-crystals with dissolving rates lower than those of the pure API while highly soluble coformers generate co-crystals with dissolving rates higher than those of the pure API.
| Co-crystal | Solventa | Solution molar ratio | P (MPa) | T (°C) | R (g g−1) | Reference |
|---|---|---|---|---|---|---|
| a THF, tetrahydrofurane.b HNA, 1-hydroxy-2-naphthoic acid. | ||||||
| 1:1 indomethacin–saccharin |
Ethanol | 1:1 |
8.0 | 50 | 0.07 | 35 |
| 1:2 theophylline–saccharin |
Ethanol | 1:2 |
8.2 | 50 | 0.12 | 35 |
| 1:1 theophylline–saccharin |
Ethanol | 1:1 |
8.3 | 50 | 0.15 | 35 |
| THF | 1:1 |
8.0 | 50 | 0.28 | 40 | |
| THF | 1:1 |
8.0 | 50 | 0.15; 0.19; 0.28 | 41 | |
| THF | 1:1 |
8.0 | 40 | 0.22 | 41 | |
| THF | 1:1 |
8.0 | 60 | 0.27 | 41 | |
| THF | 1:1 |
8.0 | 70 | 0.22 | 41 | |
| THF | 1:1 |
4.0 | 50 | 0.18 | 41 | |
| THF | 1:1 |
10.0 | 50 | 0.22 | 41 | |
| 1:1 caffeine–saccharin |
Ethanol | 1:1 |
8.0 | 50 | 0.06 | 35 |
| 1:1 sulfamethazine–saccharin |
Ethanol | 1:1 |
8.1 | 50 | 0.11 | 35 |
| 1:1 acetylsalicylic acid–saccharin |
Ethanol | 1:1 |
7.8 | 50 | 0.13 | 35 |
| 1:1 carbamazepine–saccharin |
Ethanol | 1:1 |
8.0 | 50 | 0.12 | 35 |
| 1:1 theophylline–urea |
THF | 1:1 |
8.2 | 50 | 0.09 | 41 |
| 1:1 theophylline–gentisic acid |
THF | 1:1 |
8.2 | 50 | 0.28 | 41 |
| 1:1 theophylline–salicylic acid |
THF | 1:1 |
8.0 | 50 | 0.12 | 41 |
| 1:1 theophylline–glutaric acid |
THF | 1:1 |
8.1 | 50 | 0.16 | 41 |
| 1:1 theophylline–sorbic acid |
THF | 1:1 |
8.4 | 50 | 0.26 | 41 |
| 1:1 theophylline–HNAb |
THF | 1:1 |
8.1 | 50 | 0.09 | 41 |
| 2:1 theophylline–oxalic acid |
THF | 2:1 |
8.1 | 50 | 0.13 | 41 |
| 1:1 theophylline–maleic acid |
THF | 1:1 |
8.0 | 50 | 0.13 | 41 |
| 1:1 theophylline–nicotinamide |
THF | 1:1 |
8.0 | 50 | 0.12 | 41 |
SEA is a simple, efficient and one-step method to produce co-crystals. As Padrela et al. have pointed out,41 the SEA scale-up seems relatively easy because with the exception of the high-pressure nozzle SEA consists essentially in a conventional-drying equipment which is already well established in the pharmaceutical industry.
9. Gas antisolvent crystallization, GAS
In the GAS technique compressed CO2 is added very often under stirring to a solute-containing solution in a high-pressure vessel until a desired pressure is obtained. The CO2 dissolves into the liquid solvent, causing the liquid solvent to expand, its solubilizing power to decrease and the solute to crystallize. GAS has been very often applied to obtain pharmaceutical particles.16 The co-crystals obtained by GAS and the experimental conditions used are summarized in Table 5. Shikhar et al. used for the first time the GAS method to prepare pharmaceutical co-crystals.36 An ethanol solution containing carbamazepine and nicotinamide in amounts corresponding to the co-crystal stoichiometric molar ratios 1:1 or 1:2 was introduced in a reactor and CO2 was pumped at 11.0 MPa and 40 °C and a flow rate of 90–95 mL min−1. CO2 was exhausted at regular intervals and the pressure was maintained above 7.5 MPa. 6–7 cycles of 200 mL were performed. DSC diagrams indicated that a pure 1:1 carbamazepine–nicotinamide co-crystal was obtained for the solution with a 1:1 molar ratio while the 1:1 co-crystal and the crystals of the nicotinamide in excess were obtained for the solution with a 1:2 molar ratio. SEM images showed needle shaped co-crystals, a morphology and size more favourable to dissolution than those of pure carbamazepine particles that exhibited an irregular form and dimensions exceeding the width of the co-crystal needles. Dissolution rates improved by a factor of 2.5 with respect to those obtained for pure carbamazepine.
| Co-crystal | Solventa | Solution molar ratio | T (°C) | P (MPa) | CO2 introduction rate (g min−1) | S (rpm) | Reference |
|---|---|---|---|---|---|---|---|
| a THF, tetrahydrofurane; DMSO, dimethylsulfoxide.b Three different concentrations were used. | |||||||
| 1:1 carbamazepine–nicotinamide |
Ethanol | 1:1 |
40 | 11.0 | 59–63 | — | 36 |
| Ethanol | 1:2 |
40 | 11.0 | 59–63 | — | 36 | |
| 2:1 itraconazole–succinic acid |
THF | 1:2.40 |
40 | 10.3 | — | — | 37 |
| 2:1 itraconazole-L-malic acid |
THF | 1:5.25 |
40 | 10.3 | — | — | 38 |
| 2:1 naproxen–nicotinamide |
Acetone | 2:1 |
35 | 10.0 | 2–20 | 63–500 | 39 |
| Acetone | 1:2 |
35 | 10.0 | 3 | 63; 200 | 39 | |
| Acetone | 1:2 |
35 | 10.0 | 4 | 500 | 39 | |
| Acetone | 1:1b |
35 | 10.0 | 3 | 60 | 39 | |
| Acetone | 3:1 |
35 | 10.0 | 2 | 60 | 39 | |
| Acetone | 2:1b |
37 | 10.0 | 20 | 500 | 43 | |
| Acetone | 2:1 |
37 | 10.0 | 3 | 500 | 43 | |
| 1:1 4-aminosalicylate–nicotinamide |
DMSO | 1:1 |
36 | 11.0 | 20 | 500 | 42 |
Ober and Gupta used the GAS method to prepare itraconazole–succinic acid co-crystals.37 Fig. 8 shows a scheme of the GAS setup used. The crystallization vessel is equipped with multiple sapphire windows for observation. A 0.2 μm filter is attached to the CO2 inlet line within the crystallization vessel which allows CO2 to be sprayed through the liquid solution thus enhancing mass transfer. A second vessel connected in series prior to the crystallization vessel serves as a CO2 reservoir. Temperature is controlled in both vessels. The outlet line to valve V3 is equipped with a 0.5 μm frit to prevent loss of particles during flushing. A filtered tetrahydrofurane solution containing itraconazole and succinic acid is injected in the crystallization vessel. Then CO2 is introduced into this vessel from the CO2 reservoir heated at 40 °C at a controlled flow through valve V2. Pressure is then increased at constant rate until a final pressure of 10.3 MPa is achieved. For solvent removal and CO2 flushing, valve V3 is opened and a back pressure regulator is used to maintain the desired pressure. After flushing with additional CO2, the vessel is depressurized and the co-crystals are collected.
 | ||
| Fig. 8 Schematic representation of the GAS apparatus used by Ober and Gupta:37 (P) pressure measurement, (V) valve, (TC) temperature controller, (BPR) back pressure regulator. | ||
The starting amounts of itraconazole and succinic acid in the solution used by Ober and Gupta37 were in the molar ratio 1:2.4 despite the co-crystal stoichiometry of 2:1. By using excess succinic acid the amount of unco-crystallized itraconazole, the more expensive compound, was minimized. However, this molar ratio led to the presence of excess unco-crystallized material in the final co-crystal powder. A parallel co-crystallization using heptane as a liquid antisolvent was carried out and gave a product yield of 69.4% while a product yield of 75.4% was obtained for GAS. Co-crystals of similar crystallinity, size and morphology, and identical chemical structure were produced by the two methods. However, the liquid antisolvent co-crystals exhibited an agglomeration tendency that seems to be responsible for a lower dissolution rate in comparison to that shown for the GAS co-crystal. Ober et al.38 also used GAS following a similar experimental procedure to prepare itraconazole-L-malic acid co-crystals. The HPLC analysis and the comparison of the powder XRD to that previously obtained for a single crystal indicated that the powders produced likely contain excess amorphous pure coformers in addition to the 2:1 itraconazole-L-malic acid co-crystal. The GAS co-crystals were also compared to those obtained using heptane as an antisolvent. SEM images reveal that both methods produce a homogeneous co-crystal phase with different morphology from those of the pure components. The liquid antisolvent particles had an irregular shape and smaller size than those produced by GAS. The fibrous GAS particles formed spherical aggregates and exhibited better dissolution rates than those obtained from heptane. Moreover, the GAS particles showed much better dissolution rates than the physical mixture of itraconazole and L-malic acid or the commercial itraconazole alone.
The group of Prof. Subra-Paternault obtained in 2013 (ref. 39) the 2:1 naproxen–nicotinamide co-crystal using GAS and continued to use this co-crystallization technique to the present.42,43,45 The rest of this section is devoted to the findings of this group. Their GAS apparatus presents some differences with respect to that used by Shikhar et al.36 or Ober et al.37,38 Fig. 9 shows a scheme of the GAS setup used by Subra-Paternault and coworkers.19 The acetone solution containing the two coformers is introduced into the precipitation vessel and CO2 is added. The vessel is equipped with a magnetically driven impeller whose end fitted with a turbine is plunged into the solution to allow the dispersion of the antisolvent CO2. The temperature of the vessel is controlled and kept at 35 °C. CO2 is introduced using a pump and a pressure of 10.0 MPa is attained. After precipitation, the CO2-solvent solution is drawn down at the vessel bottom and fresh CO2 is introduced in the vessel to maintain the pressure. A filter is used to hold the produced particles while the CO2-solvent solution is evacuated. Finally, the vessel is depressurized through the exit line and particles are collected.
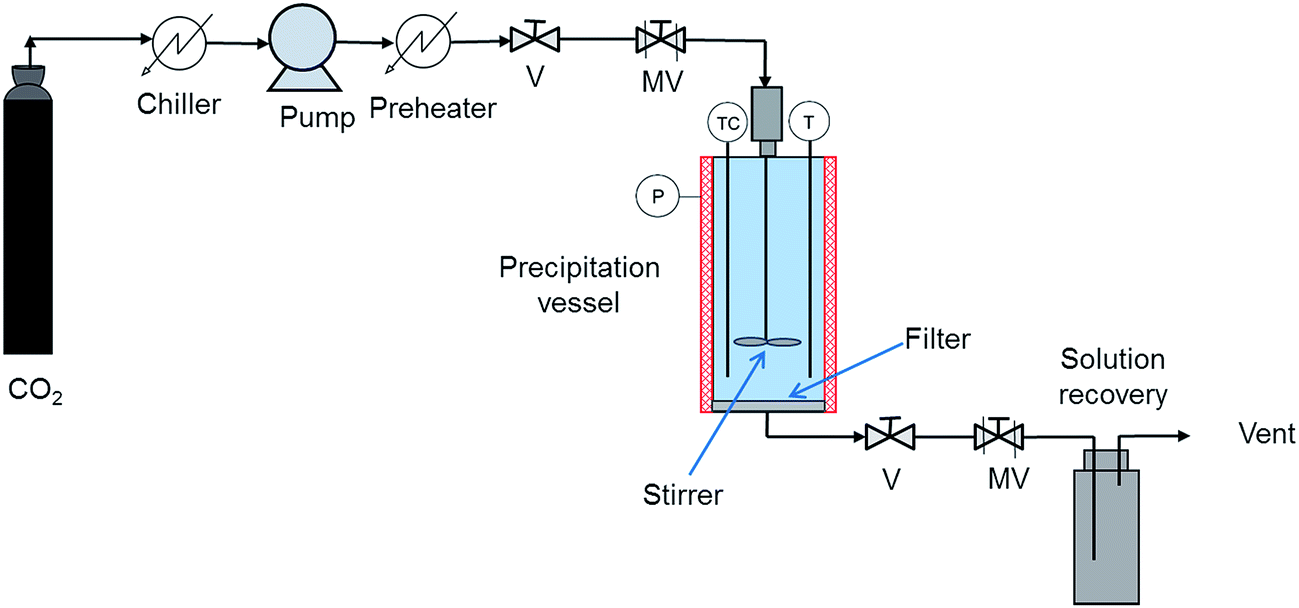 | ||
| Fig. 9 Schematic representation of the GAS apparatus used by Subra-Paternault and coworkers:19 (V) valve, (MV) metering valve, (TC) temperature controller, (P) pressure measurement, (T) temperature measurement. The stirrer consists in a magnetically driven impeller whose end is fitted with an eight-bladed disc turbine. | ||
In the preparation of the 2:1 naproxen–nicotinamide co-crystal,39 the effect of the CO2 and the initial mixture composition (naproxen:nicotinamide molar ratio) and of CO2 and acetone mixing conditions (CO2 introduction rate, stirring speed) was investigated. The purity of co-crystal powers was evaluated by HPLC and homocrystal identification was carried out by XRD analysis. Results indicated that the co-crystal purity was closed to 98 ± 2% for the initial molar ratios in solution of 3:1, 2:1 and 1:1 but levelled down to 67% when a 1:2 solution was used. The mixing conditions did not influence the co-crystal stoichiometry or purity but influenced the precipitation yield and size distribution. Agitation was found to improve the size distribution with a pronounced effect at high CO2 introduction rate. At the best conditions, a solution of naproxen and nicotinamide following the co-crystal stoichiometric ratio was used to collect 62% of the initial processed amount in the vessel as a powder of sizes below 180 μm of 2:1 naproxen–nicotinamide co-crystals with a purity of 98%. The hypothetical triangular phase diagrams of the naproxen–nicotinamide–acetone system and the naproxen–nicotinamide–(CO2 + acetone) system served Neurohr et al.39 to establish a relationship between the presence of homocrystals and the solubility of the component in the CO2–acetone mixture so that when the excess of one of the co-crystal coformers is lower than the solubility limit, it is flushed out during the continuous step of the process, whereas when it is above the limit, the compound precipitates independently. In order to check this hypothesis, Revelli et al.53 measured the solubility of naproxen, nicotinamide and their 2:1 molar ratio mixtures in CO2 + acetone mixtures at 25.0 and 37.5 °C and 10 MPa over all range of CO2 compositions. The addition of CO2 to saturated solutions produces the solute precipitation over all the range of CO2 compositions. For the quaternary system of naproxen–nicotinamide–acetone–CO2 with an initial mixture of naproxen and nicotinamide in the 2:1 molar ratio, the solid phase is composed only of the 2:1 naproxen–nicotinamide co-crystal. In these two studies,39,53 the S-enantiomer of naproxen was used since it is the one providing the desired physiological activity.
Recently, Neurohr et al.43 investigated the GAS co-crystallization of naproxen racemic mixture and nicotinamide dissolved in acetone and obtained a novel racemic co-crystal containing both enantiomers of naproxen linked to nicotinamide, the achiral coformer. The structure of the molecular complex and its intermolecular interactions were thoroughly investigated. The processed solutions had a 2:1 naproxen–nicotinamide molar ratio. The antisolvent feed rate during the pressurization step was found to have a direct influence on the co-crystallization outcome. The racemic co-crystal was obtained at slow and moderate CO2 feed rates (2 and 11 g min−1), while very fast introduction of CO2 (20 g min−1) resulted in the formation of a mixture of chiral co-crystals (conglomerate). The racemic phase was shown to be the stable form. This was the first time that a conglomerate of co-crystals with nicotinamide was obtained. All powders produced were co-crystal pure, no significant excess of naproxen or nicotinamide homocrystals was detected.
Harscoat-Schiavo et al.42 also studied the influence of isomerism on the GAS recrystallization of aminosalicylate (ASA) and its co-crystallization with nicotinamide. The experimental conditions are described in Table 5. The isomers 3-ASA, 4-ASA and 5-ASA were recrystallized as single species and important changes in the morphology were observed. In the case of 3-ASA the crystal lattice was also modified and a new polymorph was obtained. Co-crystallization of each isomer with nicotinamide resulted in the production of an ASA–nicotinamide co-crystal only in the case of 4-ASA. This co-crystal has the same stoichiometry and hydrogen networking than that previously obtained by conventional methods.
The GAS technique shares with the SAS technique the advantage of not requiring a good solubility of the API and the coformer in scCO2 while reducing the use of organic solvents with respect to traditional solution-based processes. Moreover, solvent-free particles with narrow size distributions are obtained and the organic solvent may be recovered by simple depressurization. In the GAS co-crystallizations carried out by Shikhar et al.36 and the group of Prof. Subra-Paternault,39,42,43 co-crystals of high purity are obtained. Therefore, it seems that the presence of other components together with the GAS co-crystal powders of itraconazole–succinic acid and itraconazole-L-malic acid could be avoided by modifying either the concentrations of the API and the coformer in the solution or other parameters of the GAS process. In addition, the group of Prof. Subra-Paternault has explored promising GAS applications in the co-crystallization of the API racemic mixture and the coformer, and the influence of the API isomerism on the GAS co-crystal. The group also developed a GAS mathematical model45 that will be discussed in the next section.
10. Modeling of co-crystallization using scCO2
Considerable effort has been devoted over the last years towards the understanding of the different supercritical crystallization techniques.16,55–58 However, the formation of a co-crystal using scCO2 as a solvent, antisolvent or spray enhancer requires a new approach. The co-crystal formation is based on directional molecular recognition events leading to a supramolecular structure. We need to understand how these events take place in the different supercritical co-crystallization processes. Thermodynamics, kinetics, nucleation and particle growth need to be studied.As described in the preceding sections, Prof. Gomes de Azevedo and coworkers have made efforts to understand different aspects of the various co-crystallization techniques used by this group.34,35,40,41,44 The CSS, SAS and AAS methods were simultaneously applied to obtain indomethacin–saccharin co-crystals and their differences and analogies were discussed.34 Based on a previous study for one-component crystals,54 a spraying drying mechanism was proposed for the AAS indomethacin–saccharin co-crystallization. The relation between the SEA coprecipitation mechanism and the critical pressure of the CO2 + solvent mixtures formed was established; furthermore, the influence of the SEA process variables (the nature of the coformer, the concentration of API and coformer, the value of pressure before the nozzle, the temperature in the mixing chamber) on the size distribution was also studied.35,40,41 A similar study was carried out for the CSS method.44
As to the GAS method, the modeling efforts and the thermodynamic data obtained by the group of Prof. Subra-Paternault39,42,43,53 have been already described in Section 9. Special attention should be given in this section to the GAS mathematical model developed by this group,45 the first of its kind in the case of co-crystallization. Although less frequent, the complete simulation of SAS and GAS processes including hydrodynamics of mixing, solubility and kinetics of nucleation and particle growth has been developed.59–64 Even though none of these studies is devoted specifically to the case of pharmaceutical crystals, their findings can be applied for the homocrystal production. The model proposed by Erriguible et al.45 describes the GAS co-crystallization with the aim of estimating the key parameters related to nucleation and growth and is based on the solubility data of naproxen, nicotinamide and their 2:1 molar ratio mixtures in CO2 + acetone mixtures previously obtained by Revelli et al.53 The liquid–vapour equilibrium of CO2 + acetone was modelled using the Peng–Robinson equation of state with quadratic mixing rules and its effect on the equilibrium concentration of naproxen and nicotinamide in the liquid phase was taken into account. On the other hand, the particle formation via primary and secondary nucleations and the co-crystal growth driven by diffusion were modelled on the basis of previous results for the GAS formation of the 2:1 naproxen–nicotinamide co-crystal.39 Four additional GAS experiments for the same co-crystal at a temperature of 37 °C, a pressure of 10.0 MPa, and stirring rate of 500 rpm were carried out at varying values of concentration and CO2 introduction rate in order to obtain particle size distributions. The 2:1 molar ratio in solution was used in all cases. The yield varied from 60 to 72%. A minimization algorithm allowed the estimation of the nucleation and growth parameters. The supersaturation ratio was expressed as a function of the solubility product. A good agreement was found between predicted and experimental size distributions. Nuclei formation was shown to occur mostly through secondary nucleation. The effect of concentration and CO2 introduction rate on both the crystal size and the kinetic parameters was analyzed. The initial solution concentration was shown to have little influence on crystal size. However, a relatively low introduction rate of 3 g min−1 led to bigger particles exhibiting mean sizes of ca. 50 μm while mean sizes in the 25–30 μm interval were obtained for introduction rates of 20 g min−1. Erriguible et al. attributed this behavior to a lower supersaturation and longer growth time. This successful co-crystallization model represents a very important contribution to our understanding of the application of the GAS process in the case of co-crystals.
Very recently49 the group of Prof. Subra-Paternault has also developed a model for SAS co-crystallization that is based on their earlier study dealing with single species SAS precipitation62 and the GAS co-crystallization model discussed in the preceding paragraph.45 The experimental investigations described in Section 5 were focused on the effect of CO2 and solution flow rates; their mass ratio was varied from 5 to 36. The jet disintegration and the efficiency of the mixing were not modified in this range of mass flow ratios. Consequently, the appearance of homocrystals in the SAS preparation of the 2:1 naproxen–nicotinamide co-crystals seems to be driven by thermodynamics. An innovative representation of supersaturation mapping through histograms of fluid volumes/saturation model was developed for the three species that may precipitate: the API, the coformer and the co-crystal. Experimental results seem to indicate that conditions that create discrepancies of supersaturation levels and volumes between the three species favor homo-crystallization while conditions leading to more similarities tend to favor co-crystallization. Therefore, a richer environment in solvent provides better conditions for the co-crystal production.
11. Conclusions and future perspectives
A variety of pure co-crystals have been prepared using different supercritical techniques. The use of scCO2 in the co-crystallization of pharmaceuticals reduces the thermal and mechanical stress on the API compared to grinding processes and also either eliminates or reduces the used of organic solvents compared to traditional solution methods. If a solvent is required, depressurization allows its recovery and easy recycling. Operation at moderate temperatures in an inert atmosphere avoids the product degradation. Moreover, most supercritical co-crystallization methods are one-step processes that eliminate the need of further purification steps. The solvent-free co-crystals are also micronized. This can be an inconvenient with respect to structure elucidation but presents advantages with respect to the drug dissolution profiles. These profiles are improved with respect to those of the API alone or co-crystals prepared by other methods. Nevertheless, the structure and interactions of co-crystals obtained using scCO2 are the same as those of co-crystals previously prepared by a solution or mechanochemical method. With rare exceptions co-crystals are first prepared using a conventional method and the application of a supercritical method comes later. It is a challenge to reverse this order and use supercritical methods as a mean to investigate new co-crystals. DSC and FTIR may help to identify the new structure. Next, a solution method will serve to obtain the single crystals required for structure elucidation.The possibility of tuning the fluid properties through changes in temperature and pressure that enable control of particle size and/or morphology has been demonstrated in some cases such as SEA: mean particle sizes increased as temperatures rise from 40 to 70 °C.41 These trends should be further explored.
Co-crystal purity is an important issue. It is important to make sure that traces of homocrystals are not present. This seems to be the case of most co-crystals obtained through the supercritical techniques described in this paper. In addition, the product yield is an important parameter for scale up purposes. Unfortunately, very often the yield is not reported. The values mentioned in this review indicate that good recovery ratios are obtained.
Knowledge of the solubility of the API and the coformer in scCO2 allows a better design of the co-crystallization process. Fortunately, solubility data are usually available for the binary systems CO2 + API and CO2 + coformer. These data may be used to estimate the ternary data for CO2 + API + coformer. The study of the quaternary system formed by the API, the coformer, the solvent and CO2 is more complex and requires a considerable experimental effort. Phase diagrams such as those measured by Revelli et al.53 are essential to better understand the thermodynamics of co-crystallization. However, this type of data is extremely scarce. Co-crystallization modeling efforts embracing both thermodynamic and kinetic aspects may be classified as phenomenological (those based on studying the influence of the process parameters on the characteristics of co-crystal particles produced) and mathematical. Several studies of the first kind are available. The GAS mathematical model described in the previous section45 should be recognized as a pioneering study of the second kind.
The generation of co-crystals from a racemic mixture using GAS opens a promising line. Neurohr et al.43 have shown the versatility of this technology by switching from the production of a racemic co-crystal to the formation of a mixture of chiral co-crystals (conglomerate) simply by varying the CO2 flow rate.
Another area to explore is the possible manipulation of the molecular recognition events that occur during co-crystal formation by analogy with the many examples of crystal polymorphic control reported in the literature for APIs crystallizations using scCO2. A few well documented examples of co-crystal polymorphism are found in the literature1 for conventional co-crystallization methods and it would be interesting to explore the possibilities of the supercritical methods in this context.
The energy consumption required in scCO2 experiments, the product yield that can be lower than that of a conventional co-crystallization and the initial cost associated to the application of supercritical methods in pharmaceutical industries may be mentioned as disadvantages. However, pharmaceutical companies are urged to develop production processes with very low environmental impact, fewer steps and a reduced used of organic solvents and the number of supercritical processes used in the industry is steadily increasing in the last years. In conclusion, this review shows the potential of several supercritical methods for the design and preparation of pharmaceutical co-crystals, the fast development of this line of work is anticipated.
12. The application of co-crystallization methods based on supercritical CO2 in other fields
The co-crystallization methods based on supercritical CO2 may be also applied to obtain organic co-crystals. Compared with the one-component organic single crystal, organic co-crystals present advantages similar to those of pharmaceutical co-crystals: novel and unique properties derived from the new solid structure. Organic co-crystals provide a strategy for the synthesis of novel multifunctional materials with a wide range of applications.65–67 The interactions involved in these co-crystals are classified into four categories: charge transfer, π–π interaction, halogen bond and hydrogen bond. Co-crystallization based upon halogen bonding usually involves iodines and sometimes bromines and has attracted much attention due to the resulting co-crystal potential applications ranging from molecular conductivity to phosphorescence.68 Moreover, a diarylethene co-crystal based on the aryl–perfluoroaryl interaction has been shown to convert light into mechanical work.69In the area of energetic materials, co-crystallization is considered a tool to achieve an acceptable compromise between a high energy output and a low sensitivity.70 To this end, a highly energetic but highly sensitive compound is co-crystallized with one that is less energetic and presents much less sensitivity. The result may significantly diminish sensitivity without impairing energetic performance. The problem is that while most APIs have polar groups able to interact thus leading to co-crystal formation most molecules involved in energetics exhibit nitro groups that are less versatile to establish interactions sufficiently strong. Nevertheless, this is an area of growing interest.70–73
On the other hand, polymeric co-crystalline forms consisting in structures were a polymeric host and a low-molecular mass guest are co-crystallized have been proposed for optical, magnetic and electric applications.74 In some cases an ordered polymer host structure is maintained after guest removal and a nanoporous crystalline form is obtained; in other cases an accurate control of the orientation of the polymer and guest molecules is achieved. Although a review of organic, energetic and polymeric co-crystals is beyond the scope of this study, it should be pointed out that all these applications may benefit from the use of the co-crystallization methods based on supercritical CO2 described in this paper.
Acknowledgements
We gratefully acknowledge the financial support of the Spanish Ministry of Economy and Competitiveness (MINECO) through the research project CTQ2013-41781-P. I. A. C. thanks MINECO for its support through a FPI grant.References
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CAS.
- H. D. Williams, N. L. Trevaskis, S. A. Charman, R. M. Shanker, W. N. Charman, C. E. Pouton and C. J. H. Porter, Pharmacol. Rev., 2013, 65, 315–499 CrossRef PubMed.
- S. Domingos, V. André, S. Quaresma, I. C. B. Martins and M. F. Minas da Piedade, J. Pharm. Pharmacol., 2015, 67, 830–846 CrossRef CAS PubMed.
- US GRAS List, http://www.fda.gov/food/ingredientspackaginglabeling/gras/default.htm.
- A. S. Sinha, A. R. Maguire and S. E. Lawrence, Cryst. Growth Des., 2015, 15, 984–1009 CAS.
- A. O. L. Evora, R. A. E. Castro, T. M. R. Maria, M. T. S. Rosado, M. R. Silva, A. M. Beja, J. Canotilho and M. E. S. Eusébio, Cryst. Growth Des., 2011, 11, 4780–4788 CAS.
- O. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889–1896 RSC.
- L. Wang, B. Tan, H. Zhang and Z. Deng, Org. Process Res. Dev., 2013, 17, 1413–1418 CrossRef CAS.
- A. O. L. Évora, R. A. E. Castro, T. M. R. Maria, M. R. Silva, J. H. ter Horst, J. Canotilho and M. E. S. Eusébio, Int. J. Pharm., 2014, 466, 68–75 CrossRef PubMed.
- I. A. Cuadra, A. Cabañas, J. A. R. Cheda, F. J. Martínez-Casado and C. Pando, J. CO2 Util., 2016, 13, 29–37 CrossRef CAS.
- C. C. Sun, Expert Opin. Drug Delivery, 2013, 10, 201–213 CrossRef CAS PubMed.
- J. H. ter Horst, M. A. Deij and P. W. Cains, Cryst. Growth Des., 2009, 9, 1531–1537 CAS.
- R. A. Chiarella, R. J. Davey and M. L. Peterson, Cryst. Growth Des., 2007, 7, 1223–1226 CAS.
- H. Machida, M. Takesue and R. L. Smith, J. Supercrit. Fluids, 2011, 60, 2–15 CrossRef CAS.
- I. Pasquali and R. Bettini, Int. J. Pharm., 2008, 364, 176–187 CrossRef CAS PubMed.
- N. Esfandiari, J. Supercrit. Fluids, 2015, 100, 129–141 CrossRef CAS.
- A. Kordikowski, T. Shekunov and P. York, Pharm. Res., 2001, 18, 682–688 CrossRef CAS.
- S. P. Velaga, R. Berger and J. Carlfors, Pharm. Res., 2002, 19, 1564–1571 CrossRef CAS.
- B. De Gioannis, P. Jestin and P. Subra, J. Cryst. Growth, 2004, 262, 519–526 CrossRef CAS.
- K. Moribe, Y. Tozuka and K. Yamamoto, Adv. Drug Delivery Rev., 2008, 60, 328–338 CrossRef CAS PubMed.
- A. Martín, K. Scholle, F. Mattea, D. Meterc and M. J. Cocero, Cryst. Growth Des., 2009, 9, 2504–2511 Search PubMed.
- C. Roy, D. Vrel, A. Vega-González, P. Jestin, S. Laugier and P. Subra-Paternault, J. Supercrit. Fluids, 2011, 57, 267–277 CrossRef CAS.
- M. J. Cocero, A. Martín, F. Mattea and S. Varona, J. Supercrit. Fluids, 2009, 47, 546–555 CrossRef CAS.
- C. Zhang, G. Li, Y. Wang, F. Cui, J. Zhang and Q. Huang, Int. J. Pharm., 2012, 436, 272–281 CrossRef CAS PubMed.
- L. I. Cabezas, I. Gracia, M. T. García, A. Lucas and J. F. Rodríguez, J. Supercrit. Fluids, 2013, 80, 1–8 CrossRef CAS.
- F. Zahran, A. Cabañas, J. A. R. Cheda, J. A. R. Renuncio and C. Pando, J. Supercrit. Fluids, 2014, 88, 56–65 CrossRef CAS.
- E. Badens, V. Majerik, G. Horváth and L. Szokonya, Int. J. Pharm., 2009, 377, 25–34 CrossRef CAS PubMed.
- G. Della Porta, N. Falco and E. Reverchon, J. Pharm. Sci., 2010, 99, 1484–1499 CrossRef CAS PubMed.
- E. Elizondo, S. Sala, E. Imbuluzqueta, D. González, M. J. Blanco-Prieto, C. Gamazo, N. Ventosa and J. Veciana, Pharm. Res., 2011, 28, 309–321 CrossRef CAS PubMed.
- R. T. Y. Lim, W. K. Ng and R. B. H. Tan, Powder Technol., 2013, 240, 79–87 CrossRef CAS.
- N. Murillo-Cremaes, P. Subra-Paternault, C. Domingo and A. Roig, RSC Adv., 2014, 4, 7084–7093 RSC.
- C. Vemavarapu, M. J. Mollan and T. E. Needham, AAPS PharmSciTech, 2002, 3, 29 CrossRef PubMed.
- C. Vemavarapu, M. J. Mollan and T. E. Needham, Powder Technol., 2009, 189, 444–453 CrossRef CAS.
- L. Padrela, M. A. Rodrigues, S. P. Velaga, H. A. Matos and E. G. de Azevedo, Eur. J. Pharm. Sci., 2009, 38, 9–17 CrossRef CAS PubMed.
- L. Padrela, M. A. Rodrigues, S. P. Velaga, A. C. Fernandes, H. A. Matos and E. G. de Azevedo, J. Supercrit. Fluids, 2010, 53, 156–164 CrossRef CAS.
- A. Shikhar, M. M. Bommana, S. S. Gupta and E. Squillante, J. Supercrit. Fluids, 2011, 55, 1070–1078 CrossRef CAS.
- C. A. Ober and R. B. Gupta, AAPS PharmSciTech, 2012, 13, 1396–1406 CrossRef CAS PubMed.
- C. A. Ober, S. E. Montgomery and R. B. Gupta, Powder Technol., 2013, 236, 122–131 CrossRef CAS.
- C. Neurohr, A.-L. Revelli, P. Billot, M. Marchivie, S. Lecomte, S. Laugier, S. Massip and P. Subra-Paternault, J. Supercrit. Fluids, 2013, 83, 78–85 CrossRef CAS.
- J. M. Tiago, L. Padrela, M. A. Rodrigues, H. A. Matos, A. J. Almeida and E. G. de Azevedo, Cryst. Growth Des., 2013, 13, 4940–4947 CAS.
- L. Padrela, M. A. Rodrigues, J. Tiago, S. P. Velaga, H. A. Matos and E. G. de Azevedo, J. Supercrit. Fluids, 2014, 86, 129–136 CrossRef CAS.
- C. Harscoat-Schiavo, C. Neurohr, S. Lecomte, M. Marchivie and P. Subra-Paternault, CrystEngComm, 2015, 17, 5410–5421 RSC.
- C. Neurohr, M. Marchivie, S. Lecomte, Y. Cartigny, N. Couvrat, M. Sanselme and P. Subra-Paternault, Cryst. Growth Des., 2015, 15, 4616–4626 CAS.
- L. Padrela, M. A. Rodrigues, J. Tiago, S. P. Velaga, H. A. Matos and E. G. de Azevedo, Cryst. Growth Des., 2015, 15, 3175–3181 CAS.
- A. Erriguible, C. Neurohr, A.-L. Revelli, S. Laugier, G. Fevotte and P. Subra-Paternault, J. Supercrit. Fluids, 2015, 98, 194–203 CrossRef CAS.
- K. C. Müllers, M. Paisana and M. A. Wahl, Pharm. Res., 2015, 32, 702–713 CrossRef PubMed.
- C. T. Chen, M. Tang and Y. P. Chen, Proceedings 11th International Symposium on Supercritical Fluids, 2015 Search PubMed.
- S. Hiendrawan, B. Veriansyah, E. Widjojokusumo, S. N. Soewandhi, S. Wikarsa and R. R. Tjandrawinata, Int. J. Pharm. Pharm. Sci., 2016, 8, 89–98 Search PubMed.
- C. Neurohr, A. Erriguible, S. Laugier and P. Subra-Paternault, Chem. Eng. J., 2016, 303, 238–251 CrossRef CAS.
- H. Mazen and G. Townend, US Pat., US 20080280858, WO 2008096144, EP 2117666 A1, 2008.
- G. Hofland and G. Van Rosmalen, US Pat., 2010184744, EP 2170284B1 & A2, 2009.
- L. Padrela, E. Gomes de Azevedo and S. P. Velaga, Drug Dev. Ind. Pharm., 2012, 38, 923–929 CrossRef CAS PubMed.
- A.-L. Revelli, S. Laugier, A. Erriguible and P. Subra-Paternault, Fluid Phase Equilib., 2014, 373, 29–33 CrossRef CAS.
- M. A. Rodrigues, J. Lin, L. Padrela, A. Almeida, H. A. Matos and E. Gomes de Azevedo, J. Supercrit. Fluids, 2009, 48, 253–260 CrossRef CAS.
- A. Martín and M. J. Cocero, Adv. Drug Delivery Rev., 2008, 60, 339–350 CrossRef PubMed.
- E. Reverchon and I. De Marco, J. Supercrit. Fluids, 2011, 58, 295–302 CrossRef.
- I. De Marco and E. Reverchon, Chem. Eng. J, 2011, 169, 358–370 CrossRef.
- I. De Marco, O. Knauer, F. Cice, A. Braeuer and E. Reverchon, Chem. Eng. J., 2012, 203, 71–80 CrossRef CAS.
- G. Muhrer, C. Lin and M. Mazzoti, Ind. Eng. Chem. Res., 2002, 41, 3566–3579 CrossRef CAS.
- A. Martín and M. J. Cocero, J. Supercrit. Fluids, 2004, 32, 203–219 CrossRef.
- J. Sierra-Pallares, D. L. Marchisio, M. T. Parra-Santos, J. García-Serna, F. Castro and M. J. Cocero, AIChE J., 2012, 58, 385–398 CrossRef CAS.
- A. Erriguible, T. Fadli and P. Subra-Paternault, Comput. Chem. Eng., 2013, 52, 1–9 CrossRef CAS.
- N. Esfandiari and S. M. Ghoreishi, J. Supercrit. Fluids, 2013, 81, 119–127 CrossRef CAS.
- N. Esfandiari and S. M. Ghoreishi, Chem. Eng. Technol., 2014, 37, 73–80 CrossRef CAS.
- W. Zhu, H. Dong, Y. Zhen and W. Hu, Sci. China Mater., 2015, 58, 854–859 CrossRef.
- X. Pang and W. J. Jin, Top. Curr. Chem., 2015, 359, 115–146 CrossRef CAS PubMed.
- N. D. Treat, P. Westacott and N. Stingelin, Annu. Rev. Mater. Res., 2015, 45, 450–490 CrossRef.
- G. Griffini, L. Brambilla, M. Levi, C. Castiglioni, M. Del Zoppo and S. Turri, RSC Adv., 2014, 4, 9893–9897 RSC.
- M. Morimoto and M. Irie, J. Am. Chem. Soc., 2010, 132, 14172–14178 CrossRef CAS PubMed.
- O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2011, 50, 8960–8963 CrossRef CAS PubMed.
- O. Bolton, L. R. Simke, P. F. Pagoria and A. J. Matzger, Cryst. Growth Des., 2012, 12, 4311–4314 CAS.
- K. B. Landenberger, O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2013, 52, 6468–6471 CrossRef CAS PubMed.
- Z. Yang, Q. Zeng, X. Zhou, Q. Zhang, F. Nie, H. Huang and H. Li, RSC Adv., 2014, 4, 65121–65126 RSC.
- G. Guerra, C. Daniel, P. Rizzo and O. Tarallo, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 305–322 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |