
Low temperature hydrogenation of α-angelica lactone on silica supported Pd–NiO catalysts with synergistic effect†
Pei Zhang,
Qingqing Yuan,
Li Chen,
Teng Xue,
Yejun Guan* and
Peng Wu
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Chemical Engineering, East China Normal University, North Zhongshan Road 3663, 200062, Shanghai, China. E-mail: yjguan@chem.ecnu.edu.cn
First published on 4th July 2016
Abstract
The hydrogenation of α-angelica lactone (α-AL) was achieved under mild conditions on silica supported Pd–NiO catalysts. NiO and palladium were sequentially loaded on silica by wet-impregnation and deposition–reduction, respectively. First a series of NiO/SiO2 supports with varying Ni contents were prepared by a wet-impregnation method with Ni(NO3)2 as the precursor followed by calcination in air. Then a minute amount of palladium (0.2 wt%) was loaded by a deposition–reduction method using NaBH4 as a reducing reagent. The Pd–NiO catalysts were characterized by nitrogen adsorption, XRD, H2-TPR, XPS and TEM. The NiO were heterogeneously dispersed on silica with particle sizes ranging from 10 to 50 nm, whereas Pd was finely loaded with a diameter less than 5 nm. Nanoscale intimacy between Pd and NiO was noticed by HRTEM, resulting in high catalytic activity in liquid phase hydrogenation of α-angelica lactone to γ-valero lactone (GVL) under mild conditions. 0.2Pd–9.9NiO/SiO2 showed the best activity among all the catalysts investigated, with 82% conversion and 100% selectivity to GVL within several minutes at 30 °C and 0.3–1 MPa H2 pressure.
Introduction
As a kind of promising composite material, bimetal catalysts have been intensively studied in recent years due to the well-known synergistic effect which will not be present in corresponding monometallic catalysts.1–8 From a practical point of view, the use of a bimetal catalyst is an interesting strategy as it offers many possibilities such as an increase of catalytic activity and tuning of the selectivity, especially as observed in upgrading biomass to chemicals and fuels.9–13 Precious metals, such as palladium, have been widely used in catalytic hydrogenation processes. When combined with a second transition metal, such as nickel, enhanced reactivity and/or selectivity can be achieved in many hydrogenation reactions.14–21 Nakagawa and co-workers14 reported the hydrogenation of 5-hydroxymethyl-2-furaldehyde by Ni–Pd bimetallic catalysts supported on silica (Ni/Pd = 7). The yield of 2,5-bis(hydroxymethyl)tetrahydrofuran reached 96% over bimetallic catalysts, which was more active than commercial RANEY® Ni and more selective than Pd/C. Yang et al.16 have introduced a new procedure for the reduction of aromatic nitro compounds into amines with Pd/NiO-MC (Ni-doped monolithic carbon aerogels) catalysts under ambient conditions (25 °C, 1 atm H2). Recently, Liu et al.18 reported that Pd–NiO@SiO2 nanocatalysts illustrated an excellent catalytic performance for catalytic p-chloronitrobenzene (p-CNB) hydrogenation (full conversion, 82.7% selectivity of p-chloroaniline). The conversion of p-CNB over different catalyst followed the trend of Pd–NiO@SiO2 > Pd@SiO2 > PdNi@SiO2. Synergistic effect between nickel or nickel oxide and palladium has been proposed to explain the unique catalytic behavior of these catalysts.12,18 Inspired by these studies, we herein reported the hydrogenation performance of the Pd–NiO catalyst with minute amount of Pd (0.2 wt%).α-AL has been defined as a new potential platform molecule which can be produced from lignocellulosic materials levulinic acid (LA) by dehydration (Scheme 1).22 As a perfume or food additive, α-AL is widely used in the food and tobacco industry. Moreover, it can serve as a fuel blending agent or a precursor for the preparation of γ-valerolactone (GVL), 2-methyltetrahydrofuran (2-MTHF) and levulinic acid esters. GVL is such a platform chemical that offers tremendous flexibility in downstream applications.23 It has been successfully used for the production of ionic liquids,24 butenes,25 1,4-pentanediols and alkanes,26 etc. There have been extensive studies about the hydrogenation LA and/or ethyl levulinate (EL) to produce GVL.27–34 Compared with LA or EL, α-AL can be hydrogenated smoothly under mild conditions over both Pd and Ru catalysts.22,35 Zhang et al. have reported the conversion of α-AL to GVL at 60 °C and 4.0 MPa H2 pressure using 10 wt% Pd/C catalyst in a series of ionic liquids. Among these ionic liquids, [Bmim]PF6 showed the best performance on the selectivity of GVL (99.8%). However, some ionic liquids are in solid phase at room temperature and difficult to form a homogeneous solution with the reactant as a result of low conversion. In another study, Al-Shaal et al.35 found that the GVL yield exceeded 97% with full conversion of α-AL under moderate reaction conditions (50 °C, 24 bar H2 pressure for 1 h or room temperature, 500 mL min−1 H2-flow rate for 24 h) over Ru/C catalyst. These results indicated that noble metals (Pd or Ru species) are able to saturate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds easily under mild reaction conditions. Herein, we showed that fast hydrogenation of α-AL can be achieved on a silica supported Pd–NiO catalyst with minute amount of Pd (0.2 wt%) under mild conditions.
C bonds easily under mild reaction conditions. Herein, we showed that fast hydrogenation of α-AL can be achieved on a silica supported Pd–NiO catalyst with minute amount of Pd (0.2 wt%) under mild conditions.
 | ||
| Scheme 1 The synthesis of GVL from levulinic acid via α-angelica lactone as an intermediate. | ||
Experimental
Preparation of catalysts
Catalyst characterization
Nitrogen adsorption–desorption isotherms at −196 °C were obtained on a BELSORP-Max equipment. Prior to the measurement, the samples were first degassed at 150 °C under vacuum for 6 h. Specific surface areas (SSA) were calculated by the Brunauer–Emmett–Teller (BET) method using five relative pressure points in the interval of 0.05–0.30. The pore size distribution was obtained by the Barrett–Joyner–Halenda (BJH) model applied to the adsorption isotherm.The Pd loading was quantified by inductively coupled plasma (ICP) on a Thermo IRIS Intrepid II XSP atomic emission spectrometer. About 25 mg catalysts were digested using 5 mL of aqua regia. The obtained solutions were diluted with deionized water to the desired Pd concentration.
The power X-ray diffraction (XRD) patterns were collected on a Rigaku Ultima IV X-ray diffractometer using Cu Kα radiation (λ = 1.5405 Å) operated at 35 kV and 25 mA. Transmission electron microscopy (TEM) images were taken on a FEI Tecnai G2 F30 microscope operating at 300 kV.
Temperature programmed reduction (TPR) measurements were carried out in TP-5080 (Tianjin Xianquan, China) equipped with a thermal conductivity detector (TCD). The calcined samples were treated at 100 °C for 30 min under He stream (25 mL min−1) for 1 h in order to remove the physisorbed water. Then the samples were cooled down to 25 °C in the same atmosphere, and were heated up to 630 °C at 10 °C min−1 in a 5% (v/v) hydrogen/nitrogen flow.
The X-ray photoelectron spectroscopy (XPS) was recorded on an Escalab 250xi spectrometer, using a standard Al Kα X-ray source (300 W) and analyzer pass energy of 30.0 eV. All binding energies were referenced to the adventitious C 1s line at 284.6 eV.
Catalytic tests
The catalytic hydrogenation was conducted in a Teflon-lined (60 mL) steel batch reactor. 25 mg of catalyst was loaded into the reactor along with 9.8 mL of H2O and 0.2 mL of substrate. No pretreatment on the catalysts was conducted prior to reaction. The reactor was purged with H2 for five times and then pressurized with 0.3 MPa H2. The mixture was stirred in a water-bath at 30 °C. The products were diluted with ethanol and analyzed with flame ionization detector (FID) and capillary column of DB-FFAP (30 m length and 0.25 mm internal diameter).Results and discussion
Characterization of supported catalysts
The textural properties of the supported catalysts are shown in Table 1. The surface area of SiO2 is 208 m2 g−1. As can be seen, deposition of 0.2 wt% Pd caused a slight decrease in the specific surface (from 208 to 192 m2 g−1). With the NiO loading increased (from 0.26 to 19.9 wt%), the surface area reduced substantially (from 197 to 149 m2 g−1). These can be explained by the introduction of large amount of NiO particles into the SiO2 pores.| Catalysts | SSA (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|
| SiO2 | 208 | 0.8 |
| NiO | 5.5 | 0.078 |
| Pd–SiO2 | 192 | 0.65 |
| Pd–0.26NiO/SiO2 | 197 | 0.64 |
| Pd–2NiO/SiO2 | 183 | 0.67 |
| Pd–9.9NiO/SiO2 | 166 | 0.55 |
| Pd–19NiO/SiO2 | 149 | 0.44 |
Fig. 1 shows the XRD patterns of Pd–xNiO/SiO2. The diffraction peaks located at 37, 43, 63, 75, and 79° are assigned to the NiO phase [JCPDS 44-1159]. The intensity of these peaks became weakened with the decrease of NiO loading. In all cases, no Pd diffraction peaks were observed, meaning that Pd nanoparticles were highly dispersed on metal supports or the diffraction intensity was beyond the detection limit at very low Pd loading (0.2 wt%).
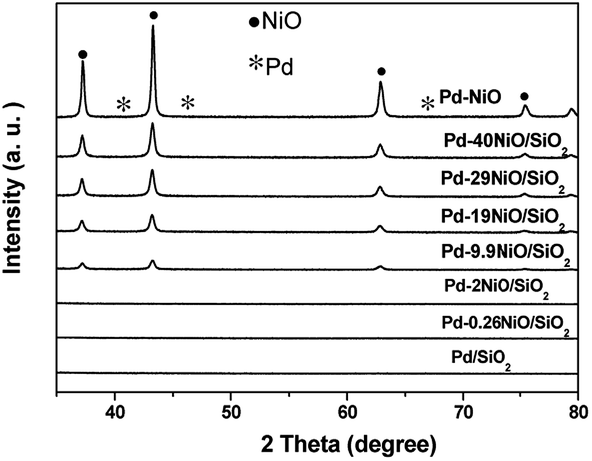 | ||
| Fig. 1 XRD patterns of Pd–xNiO–SiO2 catalysts. | ||
Fig. 2 shows the TPR profiles of the NiO/SiO2 and Pd–xNiO/SiO2 samples. The reduction profile of crystalline NiO prepared by calcining Ni(NO3)2·6H2O at 500 °C showed temperature maximum (Tmax) at 408 °C. This result is in agreement with earlier reports.37–41 9.9NiO/SiO2 and 2NiO/SiO2 presented broad reduction peak with Tmax of 388 and 418 °C, respectively. The lower Tmax of 9.9NiO/SiO2 is likely assigned to those easily reducible nickel oxide particles with large crystallites or those weakly interacting with the support.37 Pd–2NiO/SiO2 and Pd–9.9NiO/SiO2 had their Tmax at 371 °C and 381 °C, respectively. This result reveals that the presence of trace amount of Pd species enhanced the reduction of NiO. Moreover, the negative peak due to palladium β-hydride phase as commonly observed in H2-TPR profile of Pd/SiO2 (Fig. S2†) disappeared on Pd–NiO catalysts. This phenomenon certainly suggests the strong interaction between Pd and NiO, as has been reported earlier on PdFe catalysts.42
 | ||
| Fig. 2 H2-TPR profiles of NiO-bulk, 2NiO/SiO2, 9.9NiO/SiO2, 0.2Pd–2NiO/SiO2 and 0.2Pd–9.9NiO/SiO2 catalysts. | ||
We studied by XPS the surface Pd and Ni species for Pd–9.9NiO/SiO2. The Pd 3d and Ni 3p spectra were fitted and the results are shown in Fig. 3. NiO species were clearly found in Pd–9.9NiO/SiO2 according to the binding energies located at 854.7, 856.3 and 861.1 eV (Fig. 3a).43–45 Oxidized Pd2+ (Fig. 3b) species were observed for this sample (binding energies of 337.2 and 342.0 eV), which is likely due to the exposure of Pd nanoparticle in air.46 The accurate distribution of Pd and NiO on Pd–9.9NiO/SiO2 was further examined by HRTEM, high-angle annular dark field scanning tunneling electron microscopy (HAADF-STEM) (Fig. 4). From Fig. 4a and b one can see that irregular spherical particles were well dispersed on the SiO2 support. HRTEM images reveal the presence of two types of lattice spacing due to Pd nanoparticles: 0.22 and 0.19 nm assigned to Pd (111) and Pd (200) plane, respectively.18,47,48 These Pd nanoparticles were found to be closely attached on a (220) surface of NiO particles with lattice spacing about 0.14–0.15 nm.16 To get more insight on the spatial distribution of Pd and NiO, HAADF-STEM along with high resolution elemental mapping was undertaken. The mapping results indicated that NiO (shown by green dots) and Pd (shown by yellow dots) located in exactly the same zone. These results lead to the conclusion that palladium closely interacted with NiO in a big chance, which is consistent with the TPR results.
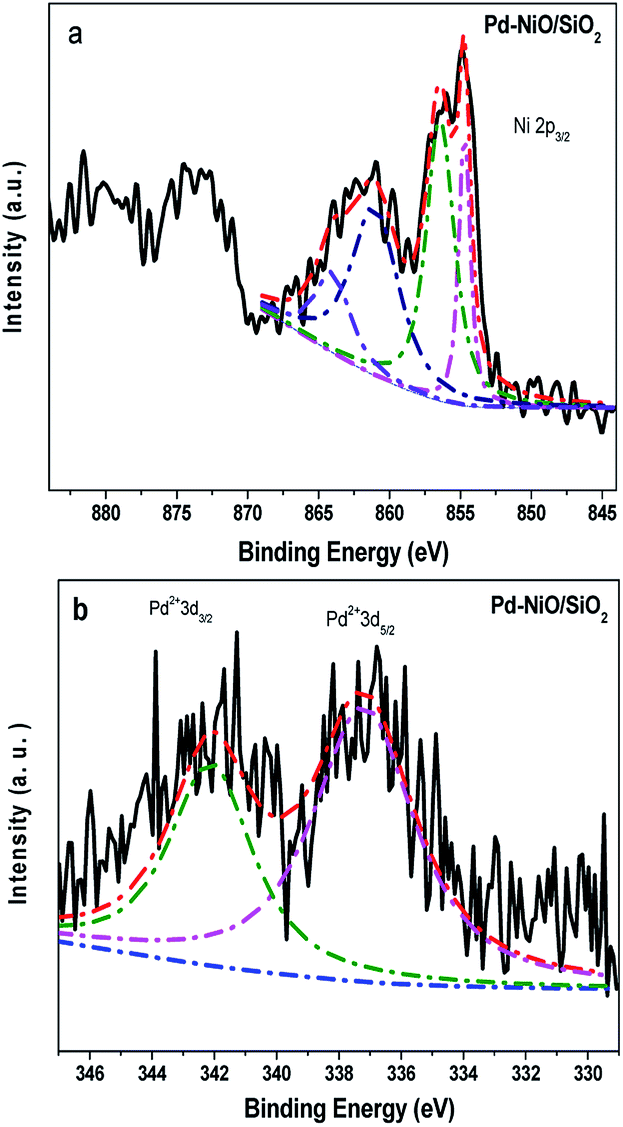 | ||
| Fig. 3 X-ray photoelectron spectra fitting of (a) Ni 2p3/2 and (b) Pd 3d of Pd–9.9NiO/SiO2. | ||
 | ||
| Fig. 4 TEM images of (a) high resolution and (b) low resolution of Pd–9.9NiO–SiO2; (c) Pd–9.9NiO–SiO2-reused; (d) HAADF-STEM-EDX mapping images of the Pd–9.9NiO/SiO2. | ||
Catalytic activities of supported Pd catalysts
Table 2 summarizes the catalytic performance of NiO/SiO2, Pd/SiO2, and Pd–NiO/SiO2 catalysts in the hydrogenation of α-AL under 0.3 or 1 MPa H2 pressure. For comparison, SiO2 (Table 2, Entry 1) and NiO–SiO2 (Table 2, Entry 2) were tested and both supports did not show any catalytic activity under 0.3 MPa H2. Increasing the H2 pressure to 1 MPa led to the production of minute amount of β-AL (Table 2, Entry 3) due to the isomerization of α-AL catalyzed by acid sites. It is likely that certain amount of acid sites related to Ni–OH species might be formed at high H2 pressure, which favors the isomerization reaction pathway. Nevertheless, NiO–SiO2 was not active at all for the hydrogenation of α-AL under mild conditions. When 0.2 wt% Pd was supported on SiO2, the α-AL conversion after 1 h was 29% with GVL selectivity being 95% (Table 2, Entry 4). This result demonstrates that Pd species were very active for the hydrogenation of CC bond in α-AL molecule even supported on an inert support such as SiO2. When supported on an active support, namely bulk NiO, the α-AL conversion was 76% after 0.5 h with enhanced GVL selectivity as 100% (Table 2, Entry 5). This result points to the synergy between Pd and NiO in hydrogenation reactions, which is most likely related to the structure modification of Pd metal by the addition of NiO.
| Entry | Catalyst | Time (min) | H2 pressure (MPa) | Conv. (%) | Sel.% | |
|---|---|---|---|---|---|---|
| GVL | β-AL | |||||
| a Reaction conditions: catalyst (25 mg), H2O (9.8 mL), α-AL (0.2 mL), 30 °C.b Physical mixture: 15 mg of NiO/SiO2 and 15 mg of Pd/SiO2.c Catalyst was pretreated with H2 at 200 °C for 10 min, cooled down to room temperature under N2 flow, and then the reaction solution was added immediately. | ||||||
| 1 | SiO2 | 60 | 0.3 | — | — | — |
| 2 | 9.9NiO–SiO2 | 60 | 0.3 | — | — | — |
| 3 | 9.9NiO–SiO2 | 60 | 1 | 1 | — | 100 |
| 4 | Pd–SiO2 | 60 | 0.3 | 29 | 95 | 5 |
| 5 | Pd–NiO-bulk | 30 | 0.3 | 76 | 100 | — |
| 6 | Pd–9.9NiO/SiO2 | 30 | 0.3 | 82 | 100 | — |
| 7 | Pd–40NiO/SiO2 | 30 | 0.3 | 78 | 100 | — |
| 8 | Pd–40Ni/SiO2 | 30 | 0.3 | 20 | 100 | — |
| 9 | NiO/SiO2 + Pd/SiO2b | 60 | 0.3 | 12 | 70 | 30 |
| 10 | Pre-reduced-Pd–9.9NiO/SiO2c | 30 | 0.3 | 27 | 100 | — |
Due to the low surface area (5.5 m2 g−1) and high cost of bulk NiO, we next tested the activity of Pd supported on NiO–SiO2 with high surface area and low Ni content. The result (Table 2, Entry 6) showed that for Pd–9.9NiO/SiO2, the conversion of α-AL after 30 min and selectivity of GVL was 82% and 100%, respectively. This reactivity was equal to or slightly higher than Pd supported on bulk NiO. As expected, the inert SiO2 with relatively larger specific surface area could disperse NiO particles very well so that more Pd–NiO active sites were provided. Further increase of NiO content did not lead to improved α-AL conversion (Table 2, Entry 7).
To clarify the role of Pd–NiO in facilitating α-AL hydrogenation, we thus performed a set of control experiments. First we tested the hydrogenation activity of physical mixture of Pd/SiO2 and 9.9NiO/SiO2 (each in 15 mg catalyst). The conversion of α-AL was 12% after 1 h with GVL selectivity of 70% (Table 2, Entry 9). This result showed that a nanoscale intimacy of Pd and NiO was the prerequisite to achieve the high hydrogenation activity of α-AL. Second we pre-reduced the Pd–9.9NiO/SiO2 in H2 flow and tested its hydrogenation activity (Table 2, Entry 10). The results clearly suggested that the reduction of Pd–NiO led to dramatic drop of α-AL conversion (27%). This unexpected result prompted us to test the catalytic activity of Pd–Ni/SiO2 catalyst in this reaction.
A series of Ni/SiO2 supports were prepared (see ESI†) by reducing the NiO/SiO2 supports in flowing H2 and then Pd was loaded via the same deposition–reduction method as Pd–NiO/SiO2 catalysts. We compared the turnover frequencies (TOF, based on α-AL conversion per Pd weight) of Pd nanoparticles supported on NiO/SiO2 and Ni/SiO2 (Fig. 5). It should be noted that in order to achieve a reliable TOF, the conversion of α-AL has been controlled to be below or close to 30%. For catalysts with high Ni loading, for instance Pd–40Ni/SiO2, the α-AL conversion was 20% after 1 h (Table 2, Entry 8), which was much lower than that of Pd–40NiO/SiO2 (78% after 0.5 h, Table 2, Entry 7). Metallic Ni significantly suppressed the hydrogenation activity of Pd nanoparticles supported thereof. Surprisingly, no significant difference in TOF between Pd–2Ni/SiO2 and Pd–2NiO/SiO2 was observed. At this stage we cannot give a convincible explanation for this catalysis behavior. It might because the influence of metal Ni on the hydrogenation activity of Pd is rather weaker comparing with that of surface NiO when Ni loading is low.
 | ||
| Fig. 5 TOF values, defined as molGVL (molPd min)−1, of Pd–xNiO/SiO2 and Pd–xNi/SiO2 in α-AL hydrogenation. | ||
Based on these results, we try to explain the pathways of α-AL hydrogenation by taking into account of previous reports.22,35 At the initial stage of the reaction, isomerization of α-AL to β-AL might occur (Table 2, Entry 3). Then, concerted adsorption of CC bond of α(and/or β)-AL and hydrogen takes place on neighbored Pd atoms and NiO surface. After the saturation of CC bonds by nearby hydrogen atoms, the formed GVL molecules desorbs to the liquid phase. It should be noted that another possible reaction mechanism cannot be excluded, wherein NiO acts as the adsorption sites of α-AL and Pd activates hydrogen. The hydrogenation is then accomplished on NiO via the H-spillover mechanism. Additionally, we tried to reveal the reaction kinetic by testing the hydrogenation performance of Pd–9.9NiO/SiO2 catalyst under different experiment conditions (Table S1†). The results showed that the hydrogenation rate was nearly zero order in lactone and first order in hydrogen. This indicated that the Pd–NiO surface was largely covered with unsaturated molecules, whereas hydrogen was only weakly adsorbed.
Catalyst reusability
The reusability of Pd–9.9NiO/SiO2 and Pd/SiO2 catalyst was evaluated through six repeated reactions. After the first run, the catalyst was separated through centrifugation, thoroughly washed and then dried at 60 °C before the next use. The activity in terms of α-AL conversion and GVL selectivity is shown in Fig. 6. It can be clearly seen that Pd–9.9NiO/SiO2 catalyst showed very stable reactivity in the first five runs and starts to deactivate in the next run. TEM image of the reused catalyst (Fig. 4c) suggested no significant aggregation of Pd nanoparticles. Elemental analysis (ICP) of the reused catalyst revealed that no leaching of Pd occurred (not shown) either. However, we noticed that the catalyst particles became badly dispersed in the aqueous solution after 6 runs, leading to strong diffusion limitations of substrates. Compared with Pd–9.9NiO/SiO2, the selectivity of GVL on Pd/SiO2 started decreasing already in the second run and decreased from 95% to 66% in the 6th run, which implying the hydrogenation activity loss of Pd in hydrogenation.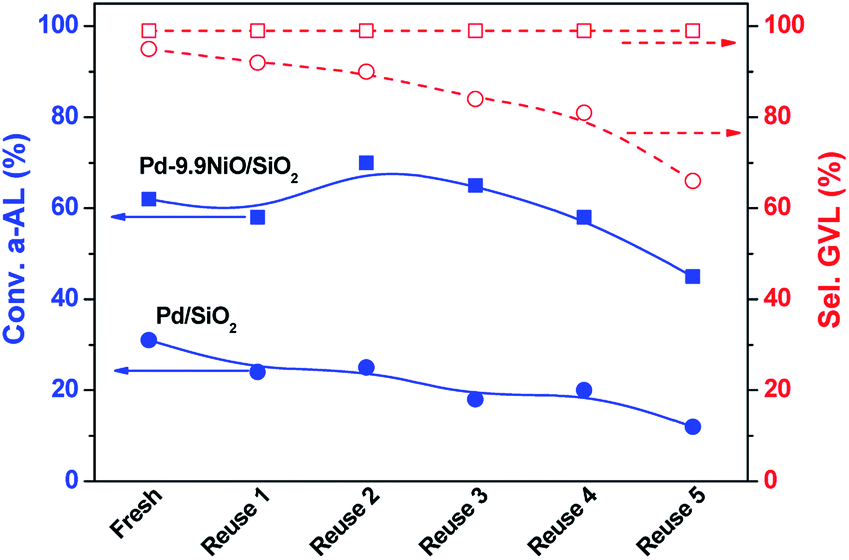 | ||
| Fig. 6 Recycle study of Pd–9.9NiO/SiO2 (Conv. (■) and sel. to GVL (□)) and Pd/SiO2 (Conv. (●) and sel. to GVL (○)) for α-AL hydrogenation. Reaction conditions: 0.4 mL of α-AL, 50 mg of catalyst, 4 mL of H2O, 30 °C, 25 min, 0.3 MPa H2. | ||
Conclusions
We demonstrated that the hydrogenation of α-AL to GVL can be successfully performed under mild conditions over a series of Pd–xNiO/SiO2 a with very low Pd loading (0.2 wt%). Amongst the catalysts investigated, Pd–9.9NiO–SiO2 was the most active one and it could be reused at least six times without Pd leaching. Pd–O–Ni species formed by the intimate interaction between Pd and NiO tend to be the active site on Pd–xNiO/SiO2 catalysts in H2 activation and the following CC saturation. The hydrogenation rate with respect to the α-AL and hydrogen pressure is nearly zero order and first order, respectively. Our results suggest that it is possible to achieve very high hydrogenation activity while using minute amount of Pd with the aid of a second cheap transition metal oxide in the conversion of biomass-derived oxygenates.
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grants No. 21203065 and 21533002).Notes and references
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075 RSC.
- C. Bernard and F. François, Coord. Chem. Rev., 1998, 178–180, 1753 Search PubMed.
- C. Bernard and F. François, J. Mol. Catal. A: Chem., 2001, 173, 117 CrossRef.
- Z. Wei, J. Sun, Y. Li, A. K. Datye and Y. Wang, Chem. Soc. Rev., 2012, 41, 7994 RSC.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099 RSC.
- F. Riccardo, J. Julius and L. J. Roy, Chem. Rev., 2008, 108, 846 Search PubMed.
- C. Liu, X. Li and T. Wang, RSC Adv., 2015, 5, 57277 RSC.
- L. Zhu, J. Zheng, C. Yu, N. Zhang, Q. Shu, H. Zhou, Y. Li and B. H. Chen, RSC Adv., 2016, 6, 13110 RSC.
- G. W. Huber, J. W. Shabaker, S. T. Evans and J. A. Dumesic, Appl. Catal., B, 2006, 62, 226 CrossRef CAS.
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222, 180 CrossRef CAS.
- J. Lee, Y. T. Kim and G. W. Huber, Green Chem., 2014, 16, 708 RSC.
- B. Chen, F. Li, Z. Huang and G. Yuan, Appl. Catal., A, 2015, 500, 23 CrossRef CAS.
- D. J. M. de Vlieger, A. G. Chakinala, L. Lefferts, S. R. A. Kersten, K. Seshan and D. W. F. Brilman, Appl. Catal., B, 2012, 111–112, 536 CrossRef CAS.
- Y. Nakagawa and K. Tomishige, Catal. Commun., 2010, 12, 154 CrossRef CAS.
- N. S. Babu, N. Lingaiah and P. S. S. Prasad, Appl. Catal., B, 2012, 111–112, 309 CrossRef.
- H. Yang, X. Cui, Y. Deng and F. Shi, ChemCatChem, 2013, 5, 1739 CrossRef CAS.
- T. Jiang, Q. Huai, T. Geng, W. Ying, T. Xiao and F. Cao, Biomass Bioenergy, 2015, 78, 71 CrossRef CAS.
- H. Liu, K. Tao, C. Xiong and S. Zhou, Catal. Sci. Technol., 2015, 5, 405 CAS.
- W. Huang, J. McCormick, R. Lobo and J. Chen, J. Catal., 2007, 246, 40 CrossRef CAS.
- Y. Huang and M. H. S. Wolfgang, J. Catal., 1999, 188, 215 CrossRef CAS.
- Y. Wang, X. Cui, Y. Deng and F. Shi, RSC Adv., 2014, 4, 2729 RSC.
- R. Cao, J. Xin, Z. Zhang, Z. Liu, X. Lu, B. Ren and S. Zhang, ACS Sustainable Chem. Eng., 2014, 2, 902 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC.
- D. Fegyverneki, L. Orha, G. Lang and I. T. Horvath, Tetrahedron, 2010, 66, 1078 CrossRef CAS.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS PubMed.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49 CrossRef CAS.
- (a) K. Kon, W. Onodera and K. Shimizu, Catal. Sci. Technol., 2014, 4, 3227 RSC; (b) F. Liguori, C. Moreno-Marrodan and P. Barbaro, ACS Catal., 2015, 5, 1882 CrossRef CAS; (c) W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657 CrossRef CAS PubMed.
- V. V. Kumar, G. Naresh, M. Sudhakar, J. Tardio, S. K. Bhargava and A. Venugopal, Appl. Catal., A, 2015, 505, 217 CrossRef CAS.
- M. Sudhakar, V. V. Kumar, G. Naresh, M. L. Kantam, S. K. Bhargava and A. Venugopal, Appl. Catal., B, 2016, 180, 113 CrossRef CAS.
- H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247 RSC.
- H. Schuette and R. W. Thomas, J. Am. Chem. Soc., 1930, 52, 3010 CrossRef CAS.
- R. V. Christian Jr, H. D. Brown and R. M. Hixon, J. Am. Chem. Soc., 1947, 69, 1961 CrossRef.
- M. Chia and J. A. Dumesic, Chem. Commun., 2011, 47, 12233 RSC.
- F. Y. Ye, D. M. Zhang, T. Xue, Y. M. Wang and Y. Guan, Green Chem., 2014, 16, 3951 RSC.
- M. G. Al-Shaal, P. J. Hausoul and R. Palkovits, Chem. Commun., 2014, 50, 10206 RSC.
- D. M. Zhang, Y. Guan, E. J. M. Hensen, T. Xue and Y. Wang, Catal. Sci. Technol., 2014, 4, 795 CAS.
- B. Mile, D. Stirling, M. A. Zammitt, A. Lovell and M. Webb, J. Catal., 1988, 114, 217 CrossRef CAS.
- N. W. Hurst, S. J. Gentry, A. Jones and B. D. McNicol, Catal. Rev., 1982, 24, 233 CAS.
- J. Zielifiski, Catal. Lett., 1995, 31, 47 CrossRef.
- G. Li, L. Hu and J. M. Hill, Appl. Catal., A, 2006, 301, 16 CrossRef CAS.
- C. W. Hu, J. Yao, H. Q. Yang, Y. Chen and A. M. Tian, J. Catal., 1997, 166, 1 CrossRef CAS.
- J. K. Kim, J. K. Lee, K. H. Kang, J. W. Lee and I. K. Song, J. Mol. Catal. A: Chem., 2015, 410, 184 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717 CrossRef CAS.
- G. Bai, H. Dai, J. Deng, Y. Liu, W. Qiu, Z. Zhao, X. Li and H. Yang, Chem. Eng. J., 2013, 219, 200 CrossRef CAS.
- F. Yu, X. Xu, H. Peng, H. Yu, Y. Dai, W. Liu, J. Ying, Q. Sun and X. Wang, Appl. Catal., A, 2015, 507, 109 CrossRef CAS.
- D. Zemlyanov, B. A. Kiss, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Havecker, A. K. Gericke, R. Schlogl, H. Gabasch, W. Unterberger, K. Hayek and B. Klotzer, Surf. Sci., 2006, 600, 983 CrossRef CAS.
- S. Dutta, C. Ray, S. Mallick, S. Sarkar, A. Roy and T. Pal, RSC Adv., 2015, 5, 51690 RSC.
- W. Wang, Y. Yang, Y. Liu, Z. Zhang, W. Dong and Z. Lei, J. Power Sources, 2015, 273, 631 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra13374f |
| This journal is © The Royal Society of Chemistry 2016 |