DOI:
10.1039/C6RA13129H
(Paper)
RSC Adv., 2016,
6, 67556-67564
New insight on facet-dependent physicochemical properties of anatase TiO2 nanostructures for efficient photocatalysis†
Received
20th May 2016
, Accepted 22nd June 2016
First published on 23rd June 2016
Abstract
Anatase TiO2, whose performance depends strongly on the exposed facets, is an efficient photocatalyst in utilizing solar energy for environmental purification. Herein, we report the synthesis of a series of TiO2 nanoparticles, with the {001} facet percentage adjusted in the range of ca. 0–100%. Comprehensive studies on the crystal structure evolution mechanisms and the corresponding influence on the physicochemical properties were conducted. Unlike previous results, we found that directly comparing the band gaps of TiO2 with different percentages of the {001} facet and the corresponding influence on the photocatalytic activity are infeasible. Experimental results indicate that 73% is the optimized {001} facet percentage for the most efficient photocatalysis. In addition to the traditional factors (e.g. crystallinity, specific surface area), this structure is also very efficient at separating the photo-carriers and adsorbing surface OH groups, which is different from previous results wherein more surface adsorbed F− ions result in less surface OH groups. This study may provide new insight on investigating the crystal facet engineering technique for efficient photocatalysis.
1. Introduction
Employing semiconductors for photocatalytic application is of significant interest because of its “green and efficient” character.1,2 Anatase TiO2, whose photocatalytic activity largely depends on the particle size, surface chemical-environment, specific surface area, morphology, and crystallinity has been widely employed and investigated.3–5 Because of the “surface science” character of photocatalysis, the influence of exposed facets on the photocatalytic activity has gained extensive attention.6–9 For anatase TiO2, new properties are expected by exposing the thermodynamic instability of crystal facets such as {001}, {110}.10–12 Several experimental studies and calculations indicate that the {001} facet is more efficient than the {101} facet for photocatalysis.13–18 The areas of application include environmental purification, hydrogen production, CO2 reduction, and solar cells.17,19,20 Some studies have attributed the higher reactive character to more five-fold coordination Ti4+ sites (Ti5c), higher surface energy, and different abilities in separating the photo-generated electrons and adsorbing the pollutants; however, they have yet to come to an agreement. Moreover, Pan et al. found that the {010}, {101}, and {001} facets have different band gaps, by measuring the absorption spectra of different particles.21 Theoretical calculations also indicate that the {001} facet has a narrower band gap than the {101} facet.13,22 It seems that more the {001} facet is exposed, the narrower band gap and higher photocatalytic activity are obtained. However, we speculate that it is not that simple, because the physicochemical properties and band gap are truly influenced by various factors, and not only the exposed crystal facets.
In the present study, we synthesized a series of {001}-faceted anatase TiO2, whose {001} facet percentage was adjusted from ca. 0 to 100. A comprehensive discussion on facet-dependent physicochemical properties and their relationships is presented, including (1) photocatalytic activities, (2) crystal growth mechanisms, (3) band gaps, (4) specific surface areas, (5) pore structures, (6) generating/separating the electron–hole pairs. This study may provide deeper insight on investigating the facet-dependent physicochemical properties of TiO2.
2. Experimental methods
2.1 Synthesis of photocatalysts
Anatase TiO2, with exposed {001} and {101} facets was synthesized by the hydrothermal method. Typically, 25 mL tetrabutyl titanate was slowly added into 15 mL of aqueous hydrofluoric acid (HF) solution (volume percentage: 0%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, and 90%) in a 60 mL Teflon-lined stainless steel vessel. The resulting mixtures changed from a white suspension to a yellow transparent solution, along with the increased HF concentration. After heating at 180 °C for 24 h, the products were washed with deionized water and ethanol, followed by drying and grinding. According to the HF volume percentage, the as-prepared samples were denoted as TF0, TF10, TF20, TF30, TF50, TF60, TF70, and TF80.
Note: (1) HF aqueous solutions with different volume percentages were prepared by changing the ratio of commercial HF (40 wt%) to deionized water. (2) No product was obtained when the HF volume percentage was 90%, thus no TF90.
2.2 Characterization
X-ray diffraction (XRD) analysis was conducted on an ARL X’TRA X-ray diffractometer using Cu Kα radiation. Morphological observations were carried out on a Hitachi S-4800 field emission scanning electron microscope (SEM). Transmission electron microscopy (TEM) analysis was conducted with a JEOL JEM-2010 electron microscope, with an accelerating voltage of 200 kV. Specific surface areas were analyzed with the nitrogen adsorption data recorded on a mMK-TriStar 3000 nitrogen adsorption apparatus (note: all the samples were degassed at 200 °C before the nitrogen adsorption measurements). X-ray photoelectron spectroscopy (XPS) measurements were conducted on a PHI5000 Versaprobe system with monochromatic Al Kα X-rays. The binding energies were referenced to the C 1s peak at 284.6 eV. UV-vis diffuse reflectance spectra were obtained on a Shimadzu 3101 spectrophotometer by employing the standard barium sulphate as the reference. Photoluminescence (PL) emission spectra were obtained on a FL3-221 fluorescence spectrophotometer (Jobin Yvon, λex = 300 nm) at room temperature. The sample usage and lamp flux were the same, so as to compare the emission intensities.
2.3 Photocatalytic activity and hydroxide radical (˙OH) analysis
RhB photocatalytic degradation experiments were carried out in a quartz reactor with circulating cooling water, and a 500 W xenon lamp as the light source. Typically, 100 mg of the photocatalyst were mixed with 200 mL RhB aqueous solution (20 mg L−1) in the reactor. Before the irradiation, the suspension was stirred for 1 h in the dark to establish the adsorption-desorption equilibrium. RhB adsorption spectra were obtained on the UV-vis spectrophotometer every 30 min after removing the suspended photocatalyst by centrifugation. ˙OH analysis was conducted with a similar procedure, based on our previous study.23
3. Results and discussion
The XRD patterns confirm the purity and anatase phase (JPCDS 21-1272) of the TiO2 samples (Fig. 1).24 With the increased HF concentration, the diffraction signal intensities of these samples are very different (Table 1). As observed, HF favors the anatase TiO2 crystal growth at the beginning, but too much HF has a negative effect on the crystallinity when its concentration reaches 80%. This can be confirmed by the lower diffraction intensity of TF80 and the disappearance of TF90. Based on the Scherrer equation and the full width at half maximum of the (101) diffraction peak, the calculated mean grain size shown in Table 1 also confirms the above mentioned discussion.25
 |
| | Fig. 1 XRD patterns of the as-prepared TiO2 samples. | |
Table 1 Physicochemical properties of the as-prepared TiO2 samples
| Sample |
HF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (mL:mL) :H2O (mL:mL) |
Mean grain size (nm) |
RCa |
RI(101):(200)b |
SBET (m2 g−1) |
Pore diameter (nm) |
| RC: Relative crystallinity, the relative intensity of the diffraction peaks to the anatase (101) diffraction peak (reference = TF0). RI(101):(200): Relative intensity, the relative intensity of (101) and (001) diffraction peaks of each sample. |
| TF0 |
0 |
7.82 |
1.00 |
1.00 |
96.74 |
12.33 |
| TF10 |
1.5:13.5 |
9.71 |
1.05 |
1.05 |
87.56 |
15.61 |
| TF20 |
3:12 |
14.06 |
1.69 |
1.69 |
79.41 |
14.48 |
| TF30 |
4.5:10.5 |
14.87 |
1.76 |
1.76 |
75.41 |
17.65 |
| TF40 |
6:9 |
17.43 |
2.23 |
2.23 |
61.60 |
14.42 |
| TF50 |
7.5:7.5 |
21.01 |
2.33 |
2.33 |
51.89 |
— |
| TF60 |
9:6 |
22.25 |
2.51 |
2.51 |
21.59 |
— |
| TF70 |
10.5:4.5 |
26.67 |
3.45 |
3.45 |
17.18 |
— |
| TF80 |
12:3 |
10.51 |
1.48 |
1.48 |
62.00 |
— |
The real mechanism on exposing the {001} facet of anatase TiO2 involves the competition for crystal growth along the [001] and [101] directions (Fig. 2a and b). To expose the {001} facet, the growth velocity along the [001] direction should be faster than in the [101] direction.13 However, our results indicate that the crystal growth along the [100] direction is also very important. The relative intensity of the (200) and (101) diffraction peaks increase from TF0 to TF70, indicating that the nanoparticles grow along the [100] direction and favor the exposure of the {001} facet. No significant change in the (004) diffraction peak may be because of the disorder accumulation of the particles during the XRD analysis, which is different from the regular TiO2 films exposed with ca. 100% ordered {001} facets.26,27
 |
| | Fig. 2 (a) Crystal growth behavior of anatase TiO2 along the [101] and [001] directions; (b) slab model of anatase TiO2 with exposed {001} and {101} facets. | |
Representative SEM (Fig. 3) and TEM (Fig. 4) analyses were conducted to support the above mentioned discussions. It was observed that the aggregated structure of TF0 is constituted by the disordered nanoparticles. The monodispersity, average particle size, and octahedral morphology of the nanoparticles are significantly improved after the addition of different amounts of HF aqueous solution. However, because of the easy adhesion of the adjacent {001} facet, high-concentrations HF will result in more serious aggregation along the [001] direction.23 This phenomenon can be confirmed by the morphologies of TF60, TF70, and TF80. For TF80, no clear plate-like morphology can be distinguished.
 |
| | Fig. 3 SEM images of the as-prepared TiO2 photocatalysts. (a) TF0; (b) TF10; (c) TF20; (d) TF30; (e) TF40; (f) TF50; (g) TF60; (h) TF70; (i) TF80. | |
 |
| | Fig. 4 TEM images of the as-prepared TiO2 photocatalysts. (a) TF0; (b) TF10; (c) TF20; (d) TF30; (e) TF40; (f) TF50; (g) TF60; (h) TF70; (i) TF80. | |
Systematical morphology changes significantly influence the structures and physicochemical properties. Therefore, we used TF0, TF10, TF20, TF40, TF60, and TF80 as the typical samples for a deeper investigation. The {001} facet percentage can be calculated based on eqn (1) and (Fig. 2b):
| |
 | (1) |
Note: θ is the theoretical angle value (68.3°) between the [101] and [001] directions of anatase TiO2.
Fig. 5 shows the typical high resolution TEM images of the samples. TF0 has an irregular morphology; even its lattice fringe cannot be observed clearly. In contrast, TF10, TF20, TF40, and TF60 all present clear lattice fringes. The lattice spacing of 0.35 nm and 0.235 nm confirm the exposure of the {101} and {001} facets, respectively.28 The much thinner TF80 shows a clear lattice spacing of 0.19 nm, which can be assigned to the (020) and (200) planes. The {001} facet percentage, which is increased along with the HF concentration, is also calculated based on more than 100 nanoparticles (Table 2). This data comes to nearly 100% for TF80, but this special structure results in low crystallinity and serious aggregation, which are negative for photocatalysis.
 |
| | Fig. 5 TEM images of (a) TF0, (b) TF10, (c) TF20, (d) TF40, (e) TF60, and (f) TF80. | |
Table 2 Physical parameters of TF0, TF10, TF20, TF40, TF60, and TF80
| Sample |
Mean edge-length a (nm) |
Mean edge-length b (nm) |
Approximate {001} facet percentage (%) |
F:Ti (%) |
–OH:O(atom) (%) |
| TF0 |
— |
— |
— |
0.00 |
7.12 |
| TF10 |
11 |
17 |
22 |
16.70 |
8.81 |
| TF20 |
19 |
24 |
40 |
18.82 |
9.85 |
| TF40 |
21 |
26 |
44 |
20.04 |
17.32 |
| TF60 |
101 |
108 |
73 |
21.79 |
36.51 |
| TF80 |
Unclear, a = b |
Unclear, a = b |
∼100 |
43.65 |
2.87 |
The surface adsorbed F atoms are the real stabilizing factor for the {001} facet (Table 2). The F 1s XPS signals, which gradually increase along with the increasing HF concentration, are mainly centered at 684 eV (Fig. 6), indicating that the F atoms are mainly adsorbed on the particle surface. In addition to the exposed {001} facet, the surface adsorbed OH is another important factor affecting the photocatalytic activity. Previous studies reported that replacing the surface adsorbed F atoms by OH groups is beneficial for photocatalysis.29 It then seems that more F atoms adsorbed on the particle surface will result in less adsorbed OH groups. However, the O 1s XPS analysis shows a different result (Fig. 7). The main peak centered at 529.5 eV and 531.5 eV can be assigned to the O atoms (Oatom) of the surface Ti–O–Ti bond and the surface adsorbed OH groups, respectively; the OH to Oatom ratio of TF0 is only 7.12%. This is gradually increased as even more F atoms are adsorbed at the same time. For TF60, about 36.51% of the O atoms originated from the surface OH. Considering the atomic structures of the {001} and {101} facets (Fig. 8), the {001} surface has larger Ti–O–Ti bond angles, while the {101} surface features lower Ti–O–Ti bond angles.13 Thus, the 2p states of the surface atoms on the {001} facets are destabilized and very reactive. On the other hand, there are 100% Ti5c atoms on the {001} facets. Both factors will result in more surface-adsorbed OH and benefit the photocatalysis. Our previous study has also pointed out that a small amount of the F atoms will also induce the formation of the Ti3+ atoms. However, the white color of all the samples indicates that the Ti3+ concentration is very low, which hardly affects the crystal structure.
 |
| | Fig. 6 F 1s XPS spectra of TF0, TF10, TF20, TF40, TF60, and TF80. | |
 |
| | Fig. 7 O 1s XPS spectra and OH/Oatom ratios of (a) TF0, (b) TF10, (c) TF20, (d) TF40, (e) TF60, and (f) TF80. | |
 |
| | Fig. 8 {101} and {001} surface structures of anatase TiO2.13 | |
Based on the above mentioned analysis, the crystal growth mechanism of the {001}-faceted anatase TiO2 is discussed. At a low HF concentration, the F atoms are primarily adsorbed on the surface and serve as the morphology control agents. Only the reactions shown in eqn (2) and (3) will happen. A lot of crystal seeds are formed and further grow into nanoparticles, exposed with the {001} facet. In contrast, a high HF concentration will dissolve some of the hydrolyzed Ti(OH)4. Reactions will undergo the processes shown in eqn (4)–(6), resulting in less crystal seeds and bigger particles. Too much HF will break the crystal growth balance, which can be confirmed by the imperfect crystalline structure of TF80 and the disappearance of TF90.
A low HF concentration:
| | |
Ti(OC4H9)4 + H2O → Ti(OH)4 + 4C4H9OH
| (2) |
| | |
(HF) + Ti(OH)4 → TiO2 + 2H2O (Crystallization)
| (3) |
A high HF concentration:
| | |
Ti(OH)4 + 4H+ + 6F− → TiF62− + 4H2O (Dissolve)
| (4) |
| | |
TiF62− + 4H2O → Ti(OH)4 + 4HF + 2F−
| (5) |
| | |
(HF) + Ti(OH)4 → TiO2 + H2O (Recrystallization)
| (6) |
Given the variations in particle size and morphology, the specific surface area was measured by nitrogen adsorption and desorption isotherms (Fig. 9a). Results (Table 1) show the specific surface area is gradually decreased from TF0 to TF70, and then improves for the ultrathin TF80. This is readily attributed to the particle size and structure evolution. Pore size distribution, which originates from nanoparticle aggregation, is also different. A value of ca. 15 nm is obtained from TF0 to TF40 (Fig. 9b and Table 1), whereas the same results are not observed for TF50, TF60, TF70, and TF80. The larger {001} facets are inclined to adhering to each other; thus, a shrinking pore structure is obtained.
 |
| | Fig. 9 (a) Nitrogen adsorption–desorption isotherms and (b) pore size distribution curves of the as-prepared TiO2 photocatalysts. | |
The band gaps estimated by the UV-vis diffuse reflectance spectroscopy (Fig. S1†) are shown in Fig. 10a. It is reported that the {001} facet has a narrower band gap than the {101} facet because of the different atomic configurations.21 It then seems reasonable that a high percentage of the exposed {001} facet will significantly narrow the band gap. However, the estimated band gap order is as follows: TF50 (3.19 eV) < TF40 < TF30 < TF20 < TF10 < TF0 < TF60 < TF70 < TF80 (3.27 eV). A gradual decrease is not observed along with the increased {001} facet percentage. VBXPS spectra reveal that the VB maxima of all the TiO2 photocatalysts are at ca. 1.96 eV, i.e., the CB minimum of the samples are different (Fig. 10b). Moreover, similar widths of the valence bands of 6.67 eV confirm similar mobility of charge carriers. Based on the discussion, we propose that it is hard to compare the band gap value of the {001} and {101} facets, based on the light absorption spectra. Various factors (e.g. particle size, defects, and physical morphology) are closely associated with the band gaps. For instance, a previous EPR study confirmed that there will be minor Ti3+ centers located in the inner structure of TF60, and the band gap can be narrowed in some contexts.30 Unlike previous reports, we consider that the band gap can only be measured and compared precisely for the same structural factors.
 |
| | Fig. 10 (a) Estimated band gaps and (b) VBXPS spectra of the as-prepared TiO2 photocatalysts. | |
Photocatalytic activities of the as-prepared samples are measured by RhB degradation under the xenon lamp irradiation (Fig. 11a). In general, improving the {001} facet percentage from TF0 to TF60 truly favors improving the photocatalytic activity. A more exposed {001} facet is favored for higher photocatalytic activity. However, a further increase in the {001} facet percentage obtains a negative effect. TF80 has the lowest photocatalytic activity among the samples; even its exposed {001} facet is nearly 100%. The fitted curves (Fig. 11b) indicate that all the reactions fit the Langmuir–Hinshelwood model, confirming the real first order degradation-reaction.31 As mentioned above, the specific surface area is gradually decreased from TF0 to TF70, indicating that the specific surface has a secondary effect on influencing the photocatalytic activity. On the other hand, the crystallinity is also gradually increased. The highest photocatalytic activity of TF60 may allow us to conclude that the optimized ratio of the {001} to {101} facet is more important in determining the photocatalytic activity.
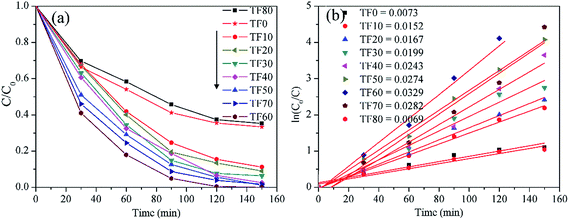 |
| | Fig. 11 (a) RhB degradation efficiency curves and (b) first order velocity constants, by employing the as-prepared TiO2 photocatalysts under the xenon lamp irradiation. | |
Photocatalytic activity is strongly correlated to the separation efficiency of photo-generated electrons and holes. In general, it is widely accepted that a lower intensity of the PL emission spectrum indicates higher efficiency in separating the electrons and holes, thus a higher photocatalytic activity can be expected.32 Recently, the “surface heterojunction” concept has also been proposed to illustrate that the co-exposed {001} and {101} facets can efficiently separate the electrons and holes. It seems that it is reasonable to obtain a lower PL emission intensity when an optimized {001} to {101} ratio is obtained. However, in the present study, different phenomena are observed (Fig. 12a). All the samples have broad PL emission spectra under the 300 nm light excitation. In addition to the band-to-band transition, there are also some peaks related to intermediary levels.33 The PL intensity decreases along with the HF introduction at the beginning, and then gradually increases from TF10 to TF70. Finally, the PL intensity of TF80 also decreases. The result is different from RhB degradation. The PL emission spectra are closely related to the photo-generated carriers, which are key factors in forming the ˙OH (eqn (7)–(11)). By employing 2-hydroxyterephthalic acid as the fluorescent molecular probe, the total amount of the generated ˙OH in different photocatalytic reaction systems are compared (Fig. S2† and 12b). Interestingly, this order is the same with the photocatalytic results, indicating that the ˙OH is a key factor in determining the photocatalytic activity.
| | |
H2O2 + O2− → ˙OH + OH− + O2
| (11) |
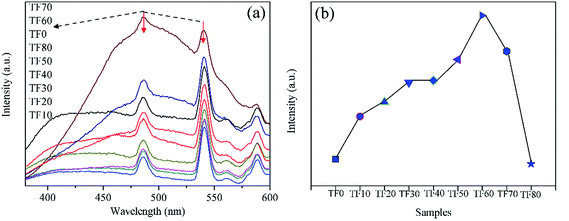 |
| | Fig. 12 (a) Fluorescence emission spectra of TiO2 photocatalysts prepared with different HF concentrations. (b) Relative amounts of the generated ˙OH in different photocatalytic reaction systems, by employing 2-hydroxyterephthalic acid as the fluorescent molecular probe. | |
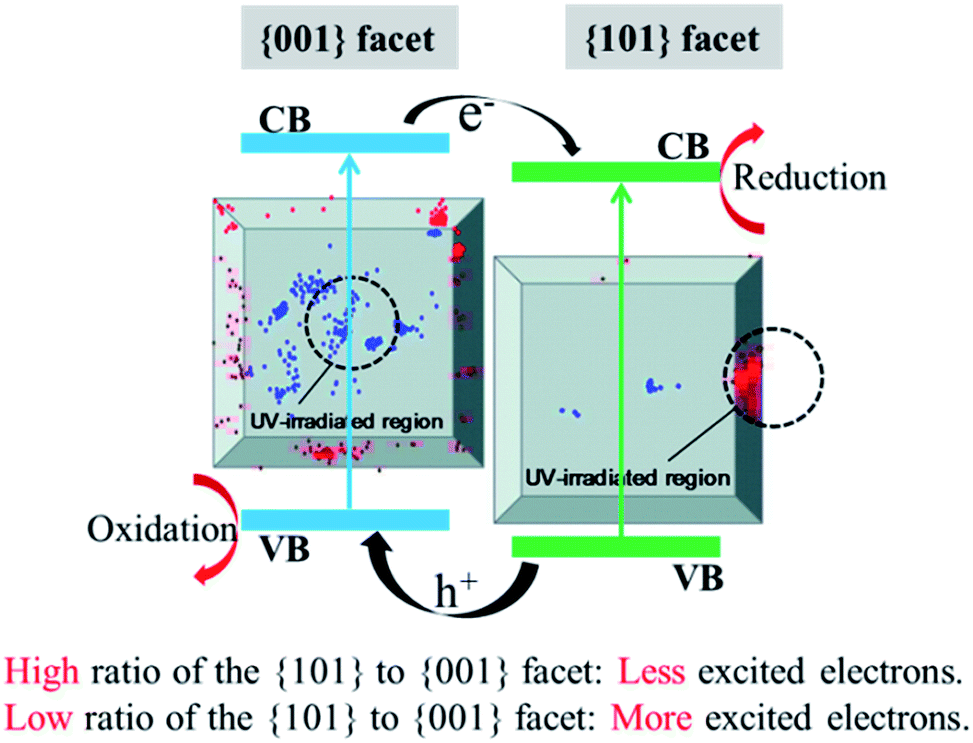
Based on the above mentioned analysis, we propose that exposing the {001} facet of anatase TiO2 is really helpful for separating the photo-generated electrons and holes. This is somewhat related to the intrinsic properties of the formed {001}-{101} surface heterojunction, which is considered a key factor in explaining the photocatalytic mechanisms of {001}-faceted TiO2 in improving the photocatalytic activities. However, this may be an unbalanced view because of the different results in Fig. 10–12. The {001} and {101} facets have different abilities in generating the electrons under the UV irradiation (Fig. 13). In general, the {001} facet excitation will generate electrons on both the {001} and {101} facets. In contrast, the {101} facet excitation mainly generates electrons on itself, but the {001} facet remains almost blank;34,35 thus, the PL emission results shown in Fig. 12a can be explained. The {001} facet percentage is gradually increased from TF10 to TF70 and more electrons and holes are generated under the UV irradiation. Therefore, the recombination of the excited carriers may also be improved, resulting in higher PL emission intensity. The lower PL emission intensity of TF80 may be due to its deficient crystal structure. On the other hand, the optimized ratio of the {101} to {001} facets is also important. In the present study, this optimal value occurs on TF60. Even more electrons and holes are generated on TF70, the less efficient {001}-{001} surface heterojunction, given its lower ability in separating the carriers and decomposing RhB. The overall consideration of the total photo-carriers and the ones available to join the photocatalytic reactions is important for designing efficient photocatalysts.
 |
| | Fig. 13 Synergistic mechanisms of different {101} to {001} facet ratios and surface heterojunctions in affecting the photocatalytic activities. Inset: the location of fluorescence bursts on the {001} (blue) and {101} (red) facets. The UV irradiation areas are inside the black circles (diameter 2 μm).35 | |
4. Conclusions
A series of {001}-faceted anatase TiO2 with different crystal structures were synthesized. Crystal growth mechanisms related to different HF concentrations have been proposed. Along with the increased HF concentration, the particle sizes along the [100] and [001] directions are gradually increased and decreased, respectively. Accordingly, the {001} facet percentage is increased to nearly 100% from TF0 to TF80. Too much HF will dissolve the small crystal seeds and then break the crystal growth balance, resulting in no TF90. Apart from the imperfect crystalline TF80, more {001} facets are favorable for generating more photo-carriers and adsorbing more F atoms, but are unfavorable for improving the specific surface area. The proper particle size and crystal structure of TF60 provide the highest ability in adsorbing surface OH groups, which can exist together with the F atoms, and separate the photo-carriers. Thus, the highest photocatalytic efficiency for RhB degradation could be achieved on TF60, which has 73% exposed {001} facets. The analysis demonstrates that the band gap, which has no direct relation to the photocatalytic performance, cannot be measured by the UV-vis absorption spectra. Our study indicates that giving consideration to various factors together, is important for designing efficient photocatalysts, and may provide new insights for understanding the crystal structures and physicochemical properties of {001}-faceted TiO2 for efficient photocatalytic applications.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 51502143), the Natural Science Foundation of Jiangsu Province (No. BK20150919, BK20130095), Key University Science Research Project of Jiangsu Province (No. 15KJB430022), and the Startup Foundation for Introducing Talent of NUIST (2014r037).
References
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668–2676 CAS.
- C. Xu, Y. Du, C. Li, J. Yang and G. Yang, Appl. Catal., B, 2015, 164, 334–343 CrossRef CAS.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- C. Li, National Science Review, 2015, 2, 143–145 CrossRef.
- G. Liu, H. G. Yang, J. Pan, Y. Q. Yang, G. Q. Lu and H. M. Cheng, Chem. Rev., 2014, 114, 9559–9612 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946–2008 RSC.
- X. Yu, B. Jeon and Y. K. Kim, ACS Catal., 2015, 5, 3316–3322 CrossRef CAS.
- T. R. Gordon, M. Cargnello, T. Paik, F. Mangolini, R. T. Weber, P. Fornasiero and C. B. Murray, J. Am. Chem. Soc., 2012, 134, 6751–6761 CrossRef CAS PubMed.
- W. K. Wang, J. J. Chen, W. W. Li, D. N. Pei, X. Zhang and H. Q. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 20349–20359 CAS.
- J. J. Chen, W. K. Wang, W. W. Li, D. N. Pei and H. Q. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 12671–12678 CAS.
- H. Xu, P. Reunchan, S. Ouyang, H. Tong, N. Umezawa, T. Kako and J. Ye, Chem. Mater., 2013, 25, 405–411 CrossRef CAS.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- W. Wang, H. Zhang, R. Wang, M. Feng and Y. Chen, Nanoscale, 2014, 6, 2390–2396 RSC.
- E. Grabowska, M. Diak, M. Marchelek and A. Zaleska, Appl. Catal., B, 2014, 156–157, 213–235 CrossRef CAS.
- L. Zhou, J. Chen, C. Ji, L. Zhou and P. O'Brien, CrystEngComm, 2013, 15, 5012–5015 RSC.
- W. Q. Wu, H. S. Rao, Y. F. Xu, Y. F. Wang, C. Y. Su and D. B. Kuang, Sci. Rep., 2013, 3, 1892 Search PubMed.
- M. M. Maitani, K. Tanaka, D. Mochizuki and Y. Wada, J. Phys. Chem. Lett., 2011, 2, 2655–2659 CrossRef CAS.
- Y. Liu, A. Tang, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2015, 137, 11327–11339 CrossRef CAS PubMed.
- S. Sun, W. Wang, D. Li, L. Zhang and D. Jiang, ACS Catal., 2014, 4, 3498–3503 CrossRef CAS.
- J. Pan, G. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- O. Lamiel-Garcia, S. Tosoni and F. Illas, J. Phys. Chem. C, 2014, 118, 13667–13673 CAS.
- W. Wang, C. Lu, Y. Ni and Z. Xu, CrystEngComm, 2013, 15, 2537 RSC.
- Q. Wu, M. Liu, Z. Wu, Y. Li and L. Piao, J. Phys. Chem. C, 2012, 116, 26800–26804 CAS.
- W. Wang, C. Lu, Y. Ni, M. Su, W. Huang and Z. Xu, Appl. Surf. Sci., 2012, 258, 8696–8703 CrossRef CAS.
- Z. Bian, T. Tachikawa, W. Kim, W. Choi and T. Majima, J. Phys. Chem. C, 2012, 116, 25444–25453 CAS.
- A. S. Ichimura, B. M. Mack, S. M. Usmani and D. G. Mars, Chem. Mater., 2012, 24, 2324–2329 CrossRef CAS PubMed.
- R. Menzel, A. Duerrbeck, E. Liberti, H. C. Yau, D. McComb and M. S. P. Shaffer, Chem. Mater., 2013, 25, 2137–2145 CrossRef CAS.
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CAS.
- W. Wei, N. Yaru, L. Chunhua and X. Zhongzi, RSC Adv., 2012, 2, 8286–8288 RSC.
- S. Gao, W. Wang, Y. Ni, C. Lu and Z. Xu, J. Alloys Compd., 2015, 647, 981–988 CrossRef CAS.
- Y. J. Yuan, Z. J. Ye, H. W. Lu, B. Hu, Y. H. Li, D. Q. Chen, J. S. Zhong, Z. T. Yu and Z. G. Zou, ACS Catal., 2016, 6, 532–541 CrossRef CAS.
- D. Li, N. Ohashi, S. Hishita, T. Kolodiazhnyi and H. Haneda, J. Solid State Chem., 2005, 178, 3293–3302 CrossRef CAS.
- T. Tachikawa, T. Ohsaka, Z. Bian and T. Majima, J. Phys. Chem. C, 2013, 117, 11219–11228 CAS.
- T. Tachikawa, S. Yamashita and T. Majima, J. Am. Chem. Soc., 2011, 133, 7197–7204 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra13129h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.