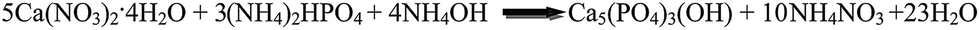DOI:
10.1039/C6RA10839C
(Paper)
RSC Adv., 2016,
6, 67565-67574
Synthesis and enhanced mechanical properties of MgO substituted hydroxyapatite: a bone substitute material
Received
27th April 2016
, Accepted 4th July 2016
First published on 11th July 2016
Abstract
Hydroxyapatite (HAp) nano-ceramic powder was synthesized successfully via microwave irradiation technique. To study the effect of MgO inclusion on the mechanical properties of pure HAp, its composites with different (0.1, 0.2, 0.3 and 0.5) wt% of MgO were prepared. The influence of sintering temperature on the mechanical properties of pure HAp and its composites with MgO was also observed at 1000, 1100, 1200 and 1300 °C respectively. Samples were characterized by using X-ray diffraction (XRD) to determine the phase stability. The mechanical properties of pure HAp and its composites with MgO were measured using several parameters such as density, Young's modulus, fracture toughness, load bearing capability and porosity. It was revealed that MgO addition significantly enhanced the grain growth as well as the mechanical properties of HAp. The HAp composite modified with 0.5 wt% of MgO sintered at 1200 °C exhibited the best mechanical characteristics. This composite exhibits density of 3.04 g cm−3, Young's modulus of 126.31 GPa, fracture toughness of 178.58 MJ cm−3 and a maximum load bearing capability of 11.61 kN. To authenticate the biocompatibility of prepared biomaterials, the cell viability (MTT assay) was carried out and the mechanically best composite was found to be compatible for biomedical applications.
1. Introduction
For several decades, research on bone mimetic biomaterials has been carried out to achieve good mechanical strength without altering the other properties. But, the challenge to prepare such a bone substitute material having comparable strength to natural bone remained almost untouched. Some metallic implants such as titanium alloys, Ti–Al alloys, stainless steel, etc. have been employed as bone substitute and they show essentially neutral in in vivo uses as permanent fixtures.1,2 Although promising for strength compared with natural bone, the metallic implantations are avoided due to the limitation of the release of toxic metallic ions. Moreover, mismatch of mechanical properties such as density and biocompatibility with natural tissues is causing the stress shielding effect.3–5 There are various approaches to develop biomimetic materials with controlled physical and mechanical properties.6 Development of such ceramic materials having appreciable mechanical strength comparable to natural bone with excellent in vivo response is also under consideration. Ceramic materials that find their clinical applications can be used to repair and reconstruct hard tissues due to long range interconnected channel which provides cell growth and healing to make these ceramics bioactive.7,8 Bioceramic material possesses unique physical properties, chemical composition and also biological response to cell adhesion, proliferation and migration.9,10 These ceramic biomaterials called bioceramic materials may be bioinert (e.g. alumina, zirconia), resorbable (e.g. tri-calcium phosphate) and bioactive (e.g. hydroxyapatite).11 Calcium phosphate minerals are found in living tissue as the most important constituents of biological hard tissues (e.g. bones, teeth, and tendons) to provide them stability and strength.12–14 At higher temperatures, HAp decomposes into secondary phases tri-calcium phosphate (β-TCP) and tetra tri-calcium phosphate (TTCP). They are the prominent stable phases at higher temperature15 but less stable and degradable in natural physiological conditions. So, they cannot be used as load bearing implants. Their degradation behavior is fully exploited in formation of bone cement. However, HAp is not only stable in in vivo conditions but also it is bioactive and promotes the bone ingrowths on implantation. So, HAp finds momentous applications in various prosthetic, dental and other clinical as well as biomedical fields. Ability to fill the missing segments in bone tissues and supports for host tissues makes it more exploitable as among tissue engineering materials.16,17
Hydroxyapatite: HAp [Ca10(PO4)6(OH)2] is chemically similar, to the crystallographic composition of inorganic phase of natural bones and teeth.18,19 Biological applications, biocompatibility, bioactive characteristics of HAp depend on many properties, structure, composition, crystallinity, surface morphology and interconnected porous structure similar to natural bones.20,21 HAp nano structured powders are highly biocompatible, osteoconductive and excellent biomaterials as bone graft, bone filler substitute and implant materials.22–24 The application of pure HAp for bone replacement is limited due to its poor mechanical strength as compared to the natural bone and teeth.25 No biomaterial could be prepared till now which can stand with strength of the order of human bone. The strength as well as other mechanical properties of HAp depends on microstructural properties such as porosity and the formation of secondary phases.26 The porosity of a biomaterial plays an important role in tissue regeneration due to pore interconnections. Porous architecture of HAp as a bone graft substitute material provides mechanical support due to natural tissue growth and no adverse reactions.27,28 The mechanical properties of porous HAp highly depend on porous architecture and size of pore.29 In order to mimic natural bone, HAp can be tailored for desired mechanical strength and degradation by substitution/doping and/or incorporation of some additives to form its composites.30–32 Since, the mechanical performance of pure HAp is very poor, a number of different biologically acceptable elements/materials (oxides and carbonates) have been added into pure HAp to form various composites of enhanced bioactivity as well as mechanical properties without altering biological properties. The different biocompatible oxide additives such as silica (SiO2),33,34 zirconia (ZrO2),35–37 alumina (Al2O3),38 reduced graphene oxide (r-GO),39 cerium oxide (CeO2),40 and manganese dioxide (MnO2)41 have been attempted to investigate their respective mechanical characteristics. A small amount of MnO2 as additive is found to be more beneficial at low temperature sintered HAp for densification and mechanical properties.41 HAp-CNT nano composite sintered by spark plasma method also promising improved mechanical properties is reported.42 Recently, Schumacher et al., reported the non-linear mechanical behavior of HAp when doped with strontium.43 More recently, we have reported a significant improvement in tribological and other mechanical properties of HAp composites with SrCO3 and ZrO2. The mechanical and biodegradable properties of MgO added composites and alloys are especially attractive for bone and teeth implant applications due to its excellent biocompatibility, high degradability, low weight and density similar to natural bones.44,45 MgO included HAp are widely used as bone graft materials due to faster recovery of host material (bone) by gradually releasing Mg ions from implanted materials.46–48 Relatively, low melting temperature of MgO offer it to fuse with HAp nanoparticles at higher temperatures during sintering, which consequences a significant enhancement in MgO assisted HAp composites.49 Previously, several studies on HAp modified by different mol/wt% of Mg2+, MgO have been carried out. The beneficial effect of MgO specially for enhancing the hardness and toughness of HAp also been reported.44–51 HAp composed of 0.5 wt% MgO, achieved more than 98% density when sintered at 1100 °C.52 However, its Young's modulus increased up to 125.90 GPa from 119.5 GPa (pure HAp) when 1.0 wt% MgO is added in HAp. The MgO inclusion into the pure HAp powder is strongly expected to affect the grain growth in HAp and improves the mechanical properties of HAp significantly.
In order to enhance the proper strength of HAp, a comprehensive study on the effect of the different amount of MgO (0.1, 0.2, 0.3 and 0.5) wt% on the mechanical properties of HAp is carried out. Firstly, pure HAp powder was synthesized via microwave irradiation technique. This technique is known to be time and energy saving, extremely efficient to obtain ultrafine and high crystalline HAp powder. Further, the MgO–HAp composites were prepared by adding estimated amount of MgO into HAp via ball milling method. Surface properties of HAp samples were characterized by various techniques such as SEM and contact angle measurements. The hydrophilic behavior of pure HAp and MgO doped HAp composites were also studied by contact angles measurements. Lower contact angle is favorable for hydrophilic materials.53 To check the biocompatibility of prepared HAp and its composites, the most acceptable testing (MTT assay) is also carried out.
2. Experimental procedure
2.1. Materials and method for synthesis
For the synthesis of pure HAp powder the calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), diammonium hydrogen phosphate ((NH4)2HPO4), ethylene diamine tetra-acetic acid (EDTA) and MgO of high purity (>99%) were purchased from Merck Pvt. Ltd., India and were used as starting raw materials. All the chemicals were used without any further purification. The following chemical reaction takes place for the formation of HAp.
Calcium nitrate tetra hydrate (1.0 M) and diammonium hydrogen phosphate (0.6 M) were separately dissolved into 100 mL of double distilled water to maintain the Ca/P molar ratio 1.67. Stoichiometric amount of EDTA (1.0 M) was used as capping agent and introduced into the 100 mL of calcium nitrate transparent solution. EDTA introduced calcium nitrate mixture was stirred by magnetic stirrer for ∼30 min and diammonium hydrogen phosphate aqueous solution poured into Ca–EDTA contained solution to form the milky white suspension of pH 4. The final pH of this suspension was kept at ∼12 by adding liquid ammonia drop wise and through continuous stirring for 2 h. The obtained solution of a certain pH was kept into a glass condenser equipped microwave oven bombarding the microwaves of frequency 2.45 GHz using 600 W power for 10 min followed by a working cycle of 5 s on and 10 s off. After 2 min of the microwave bombarding, the transparent solution started to turn once again into a bright milky suspension and the formed precipitate settled down. The precipitate was then washed and centrifuged in triplicate with deionized water to remove EDTA, NH4+, NO3− ions and other chemical impurities. The paste-like material thus obtained was then dried at 120 °C for 24 h to get bright white HAp in powder form. The HAp powder was calcined at 800 °C for 2 h for its further crystallization and removal of other impurities and residuals. In order to prepare four samples of MgO substituted HAp composites, already estimated different (0.1, 0.2, 0.3 and 0.5 wt%) amounts of MgO were added individually into a certain amount of HAp powder and were ball milled for 10 h in dry and acetone media. A certain amount of homogeneously ball milled MgO–HAp composite sample was then compacted into pellet form of size (height × diameter: 20 mm × 13 mm) by applying 2 ton pressure with a dwell of 60 s. In order to see the effect of sintering, the compacted pellets were then sintered at different temperatures was also formed and sintered in the same temperature range (i.e. 1000–1300 °C) at a constant heating and cooling rate of 3 °C min−1. This pellet of pure HAp was processed like-wise for control purpose.
2.2. Materials and methods for cell viability
In order to check the biocompatibility of the synthesized HAp nanoparticles, MTT assay was performed on LE cells. A standard solution of 1 mg mL−1 (for nanoparticles of sample 0.5 wt% MgO–HAp), in Dulbecco's phosphate-buffered saline (DPBS) or dimethyl sulfoxide (DMSO) was prepared. Dilutions of nanoparticles viz. 1, 5, 25, 50 and 100 μg mL−1 were prepared using Dulbecco's modified eagle medium (DMEM). Overnight grown LE cells in 96-well microliter plate were washed with DPBS and 100 μL of various diluted concentrations of nanoparticles were added. The plates were incubated at 37 °C in a humidified chamber with 5% of CO2 for 48 h. 100 μL of MTT solution (12.5 mg of MTT into 2.5 mL DPBS) was added to each well and incubated at 37 °C in a humidified chamber with 5% CO2 for 3 h. Later the MTT, media along with nanoparticles were removed and the cells were washed with DPBS. Then 100 μL of DMSO was added and incubated at 37 °C in a humidified chamber with 5% CO2 for 10 min. Finally the absorbance was measured at 570 nm.
2.3. Materials characterizations
In order to determine the phase formation and phase decomposition of pure HAp as well as MgO–HAp composite powders, the X-ray diffraction (XRD) patterns were recorded by using a Rigaku Miniflex-II X-ray diffractometer equipped with monochromatic Cu-Kα radiation (λ = 0.15418 nm) operated at 40 kV and 40 mA. The data was recorded in the 2θ range of 20° to 50° with scanning rate of 3° min−1. XRD patterns of the pure HAp as well as sintered MgO–HAp composites were compared with JCPDS file no. 24-0033 (standard data for hexagonal HAp) for different phases, phase dissociation and formation of the reaction product. The molecular structure and intra/inter molecular bonding between HAp and MgO–HAp were studied by using ATR-FTIR spectrometer having range from 4000–400 cm−1 (Perkin Elmer spectrum 65, FT-IR spectrometer; Perkin Elmer, M A.). The densities of pure as well as MgO–HAp samples were measured individually by using Archimedes method. Double distilled water was used as substituting material because it can easily penetrate into the pores of HAp based samples. The sintered pellets of the HAp and its composites were immersed in double distilled water; no vapors are seen coming out of the pellets. The dry, soaked and suspended weight of a pellet was measured and replicated for all the pellets. By using the following formula the bulk densities of all the sintered samples were calculated:
The mechanical properties of MgO–HAp composite samples were measured by using universal testing machine (UTM Instron 3639) in compression mode on cylindrical pellets of 1.3 cm diameter and 2.0 cm length/height. During characterization, the temperature and relative humidity were noted to be 23 °C and ∼50%. The crosshead speed was maintained at 5 mm min−1. The Young's modulus was calculated from the slope of stress–strain curve for each sample. To study the load bearing behavior of HAp as well as its composites with MgO, the load displacement curves were plotted until their failure. The fracture toughness of samples was measured by the area under stress–strain curve within limit of initial point to elastic limit point for each sample. Porosity is defined as the percentage of void space in the solid. Porosity of each sample was measured according to the equation
The surface morphologies and microstructures of fracture in HAp and MgO–HAp samples were carried out by using scanning electron microscope (JEOL JSM-6400) supported by elemental analysis using energy dispersive X-ray spectroscope (EDAX) at 20.0 kV and 15.0 mA. The Brunauer–Emmett–Teller (BET) specific surface areas and pores diameter were also tested of the pure HAp and its composites doped with MgO powder samples were measured using a Quantachrome Autosorb-3b BET Surface Analyzer. Transmission electron microscopy (TEM) was carried out to record TEM images and diffraction pattern was taken using JEOL 2100 field emission gun transmission electron microscope. For preparing the TEM samples, tiny amount of the powder was taken and then bath sonicated in isopropyl alcohol for 25 min. Some drops were then cast onto the holey carbon grids and allowed to dry in vacuum for duration of 30 min. The TEM samples were then left under vacuum for 12 h. The surface hydrophilic property of pure as well as MgO substituted HAp samples was measured using an ‘Advanced Goniometer’ (500, Rame-Hart, Inc.) was used to measure the contact angle, θ°. During contact angle measurement the sessile drop of distilled water was deposited on the surface of sample by using a micrometric syringe. The volume of sessile drop was kept constant for all every measurement of the samples. The drop was recorded when the stabilization was achieved completely and the contact angle was observed when liquid spread on sample surface. The contact angle revealed the hydrophilic behavior and chemical heterogeneity of observed solid surface for HAp and its composites. If the observed contact angle is less than 90°, it means the material is hydrophilic. However, if the liquid spread out over the wide area of sample while contact angle is greater than 90°, it indicates that the materials are hydrophobic.
To check the cell viability of the HAp based material, equal numbers of 5000 cells per well were seeded in 96-well plates and grown at 37 °C in a humidified chamber with 5% CO2 for 48 h. Cells were then treated with 1, 5, 25, 50 and 100 μg mL−1 of 0.5% MgO–HAp nanoparticles and allowed to grow for 48 h. The cell viability was assayed by MTT dye uptake. The mean absorbance at 570 nm is represented as cell viability percentage of the control and is mean ± SD of 96 wells. The values are a representative of three experiments performed independently.
3. Results and discussion
3.1. X-ray diffraction
The XRD patterns of synthesized HAp and HAp composites with different wt% of MgO sintered at 1200 °C are shown in Fig. 1. The appearance of sharp and high intensity peaks in the XRD pattern having good agreement with JCPDS file (card no. 74-0566) revealed the formation of pure and crystalline HAp samples. The XRD pattern of pure HAp sample showed no secondary phase or other impurities in the profile. The XRD patterns of MgO–HAp composites are almost similar to the XRD pattern of pure HAp except a tiny peak of MgO and two similar intense peaks of β-TCP. The appearance of characteristic peak of MgO at 36.90° indicate the presence of MgO because it is substituted into HAp and β-TCP is the secondary phase of calcium phosphate when sintered at or above 1200 °C. The careful examination of XRD results showed that increasing wt% of MgO does not alter phase stability of HAp–MgO structure.
 |
| | Fig. 1 The formation of HAp and MgO–HAp composite is confirmed by the XRD in scanning range 20–60° sintered at 1200° (a) pure HAp. (b) 0.1 MgO–HAp, (c) 0.2 MgO–HAp, (d) 0.3 MgO–HAp (e) 0.5 MgO–HAp. | |
3.2. Infrared spectroscopic analysis
To study the molecular structure of prepared samples, the IR spectroscopy was also monitored. The FT-IR absorption spectra of pure HAp and MgO–HAp samples are shown in Fig. 2. Strong absorption bands appearing in between 950 and 1230 cm−1 were the characteristic stretching bands of O–P–O in PO43− of HAp. A strong characteristic band at 970 cm−1, noticed in IR pattern of pure HAp was attributed to ν1-symmetric stretching mode of P–O in PO43−. As the MgO was incorporated, this band was blue shifted by 6 cm−1 and appears at 976 cm−1 for all the composite samples. IR bands centered at the 1030, 1080 and 1130 cm−1 are attributed to ν3 asymmetric stretching of P–O in PO43−. Vibrational mode of water is noticed at 606 cm−1 only in IR profile of pure HAp sample. A sharp and strong shoulder at 550 cm−1 may be assigned to ν4 asymmetric bending mode of O–P–O in PO43− and this band is also found to be blue shifted to 564 cm−1 (by 14 cm−1) as MgO was added. A weak shoulder appeared at 492 cm−1 in all the IR spectra is due to ν2 symmetric bending mode of O–P–O in PO43−. ATR-FT-IR characterization of the prepared samples confirmed the formation of HAp due the presence of various modes of their functional groups.
 |
| | Fig. 2 The band assignments of HAp and MgO–HAp in range of 400–4000 cm−1 sintered at 1200 °C (a) pure HAp, (b) 0.1 MgO–HAp, (c) 0.2 MgO–HAp, (d) 0.3 MgO–HAp, (e) 0.5 MgO–HAp. | |
3.3. Density analysis
Density of different MgO–HAp composites has been measured and numerical results have been shown in Table 1. Results show the density of pure HAp increases with increase in the sintering temperature; however, the density of MgO–HAp composites depends not only on sintering temperature but also on the MgO concentration. The density of MgO–HAp composites increases significantly with the MgO concentration as well as with the sintering temperature up to 1200 °C. The densities of pure HAp as well as composites except least amount of MgO added HAp sample, are found to be decreased at sintering temperature 1300 °C. In this section of study, the sintering temperature as well as concentration of MgO is optimized to get maximum density of the composites. On the basis of above results, the highest value of density 3.04 g cm−3 is determined for 0.5 wt% of MgO added HAp sample sintered at 1200 °C. In this task to enhance the density of pure HAp, incorporation of MgO and its concentration are proved to be much effective on one hand. On the other hand, sintering temperature played key role to make denser the HAp based materials.
Table 1 Effect of MgO doping on the density of HAp and MgO–HAp samples with sintering temperature, variation of contact angle, BET surface area and average pore size sintered at 1200 °C
| Composition |
Density (g cm−3) at different sintering temperature (°C) |
Contact angle (°) at 1200 °C |
BET surface area (m2 g−1) |
Average pore diameter (nm) |
| 1000 |
1100 |
1200 |
1300 |
| HAp |
2.50 |
2.75 |
2.89 |
2.94 |
21.20 |
26.72 |
12.91 |
| 0.1 MgO–HAp |
2.53 |
2.87 |
2.94 |
2.97 |
28.98 |
25.20 |
40.79 |
| 0.2 MgO–HAp |
2.63 |
2.93 |
2.98 |
2.97 |
29.15 |
24.34 |
80.45 |
| 0.3 MgO–HAp |
2.68 |
2.91 |
3.01 |
2.98 |
31.35 |
23.65 |
93.70 |
| 0.5 MgO–HAp |
2.71 |
2.98 |
3.04 |
2.97 |
40.37 |
21.87 |
111.2 |
3.4. Mechanical behavior
Young's modulus is the one of the important mechanical parameters required to study the mechanical behavior of a material. In this regard the Young's modulus and its variation with MgO concentration at different sintering temperatures are also studied and shown in Fig. 3(b). The MgO addition and its increasing wt% significantly enhanced the Young's modulus of HAp. The sintering temperature also enhanced the Young's modulus parallel up to 1200 °C and then modulus is found to be reduced. The highest value of Young's modulus 126.31 GPa noticed for 0.5 wt% MgO–HAp composite sample sintered at 1200 °C whereas, it is 114.20 GPa for pure HAp. On careful examination of density as well as Young's modulus patterns, it is observed that their dynamics comply with each other throughout the physico-chemical conditions. Both these parameters increased linearly with increasing wt% of MgO and found to be highest for 0.5 wt% MgO level in HAp at the same sintering temperature 1200 °C. Interestingly, both the physical parameters density and Young modulus shows reverse effect i.e. decreases dramatically at 1300 °C. Fig. 3(c) shows the effect of addition of MgO, increasing with wt% of MgO and sintering temperature on the fracture toughness of HAp. Effects of MgO addition, increasing wt% of MgO and sintering temperature on the fracture toughness were found to follow similar dynamics as earlier measured mechanical properties, density and Young's modulus. The MgO incorporation and its increasing wt% significantly improved the fracture toughness and observed highest (178.58 MJ cm−3) for 0.5 wt% at 1200 °C. The fracture toughness increased with increasing of sintering temperature up to 1200 °C and it decreased by further increasing the sintering temperature beyond 1200 °C. In order to authenticate the soundness of mechanical behavior of prepared composite materials, the load–displacement measurements were also carried out for thermally optimized samples sintered at 1200 °C and are shown in Fig. 3(a) similar to the previously measured mechanical properties, load bearing capability was also significantly enhanced by MgO incorporation. Load bearing capabilities of the composites were found to be increased for 1–2 wt% of MgO in pure HAp. Pure HAp showed 2.05 kN load capability. It is interesting to note that the load capacity of 3 wt% of MgO added HAp composite sample increased abruptly and a highest value of 11.61 kN was noticed for 0.5 wt% of MgO added HAp composite similar to the other mechanical properties of this sample. The addition of 5 wt% of MgO amount into the pure HAp significantly enhanced the load bearing capability of pure HAp.
 |
| | Fig. 3 Mechanical and porosity behaviors of MgO–HAp composite samples with sintering temperature, (a) load vs. displacement graph of HAp and MgO–HAp samples sintered at 1200 °C, (b) variation of Young modulus of HAp and MgO–HAp with sintering temperature, (c) dependency of fracture toughness of HAp and MgO–HAp samples on sintering temperature (d) the reduction of porosity of HAp and MgO–HAp samples with sintering temperature and porosity. | |
3.5. Microstructural behavior
The surface morphology of HAp and MgO–HAp fractured samples sintered at 1200 °C are shown in Fig. 4. In the images, it can be easily seen that MgO addition significantly affects the grain growth of HAp causing the enhanced mechanical properties. The microstructures of MgO–HAp composites show proper grain growth. The pores are represented in red colored dotted rings. The SEM image 4(a) of pure HAp sample showed pores of numerous irregular shaped and few of them are interconnected to each other. Therefore, the densities as well as other mechanical properties of pure HAp sample are poor rather than its composites. As the MgO concentration was increased, comparatively denser microstructure is obtained because MgO is a highly fusible and it diffuses into the interconnected pores and reduces the porosity. The MgO particles at 1200 °C are expected to be molten and promote densification of HAp samples. The 0.5 MgO–HAp composite exhibited highest density and least porous microstructure. Although some pores are observed because HAp decomposes into secondary phases due to dehydroxylation at higher temperature. It is observed that most of the pores were present at grain boundaries of HAp grains. Density/porosity shown in microstructures (see Fig. 4(a)–(e)) of the pure HAp as well as its composites with MgO runs parallel to the density calculated earlier for each sample. In the image 4(a) of 0.1 MgO–HAp composite, comparatively regular and homogeneous pores are observed. However, pure HAp sample showed highly porous structure and inhomogeneous pores are distributed entire image. These results are well consistent with the results of TEM. As the MgO concentration was increased (0–0.5 wt%) the particle sizes as clearly seen in images 5(b)–(e), increased due to the agglomeration of the particles and due to the interaction of HAp with MgO particles. In principle, if particle size is increasing then the voids among them will also be increases and hence the pore size is also increasing with increasing MgO concentration. The addition of MgO in HAp may be possible method to control the various properties such as mechanical, porosity, pore interconnectivity and hydrophilic nature. In the image 4(b) of 0.3 wt% MgO added HAp composite, a non-linear crack layer (see red dotted line) appears. The MgO particles (as shown by yellow arrows) acted as interlocking between the HAp grains along the crack developed.
 |
| | Fig. 4 Scanning electron microscopy images of HAp and MgO–HAp samples show porous and interconnected microstructures (a) pure HAp, (b) 0.1 MgO–HAp, (c) 0.2 MgO–HAp, (d) 0.3 MgO–HAp and (e) 0.5 MgO–HAp. | |
3.6. Energy dispersive X-ray analysis
The elemental analysis of selected SEM of pure HAp, 0.3 MgO–HAp and 0.5 MgO–HAp composites sintered at 1200 °C are shown in Fig. 5(a)–(c). The yellow color arrows were marked on the facets of the fine grains for which the EDAX were recorded. It is observed from the EDAX spectrum that well resolved peaks correspondence to different elements, Ca, P, O were present and also confirms the synthesis of pure HAp (Fig. 5(a)) and presence of the dopant of MgO. Only a single unwanted peak of Na was observed for the 0.3 MgO–HAp composite which might be due to the foreign impurity. The surface area was inversely proportional to the content of MgO (wt%) that was consistent with porosity and surface area measurements (Fig. 6(a)). The average pores sizes diameter were inversely proportional to the content of MgO (wt%) that was consistent with porosity and surface area measurements. The surface area increases with increase in the concentration of MgO (wt%) (Fig. 6(b)). The values of the surface area and pore size of the synthesized MgO-doped HAp lie in the range from 21.87 to 26.72 m2 g−1 and 12.91 to 111.2 nm, respectively (Table 1).
 |
| | Fig. 5 EDAX spectra of the pure HAp and its composites doped with MgO: (a) pure HAp, (b) 0.3 MgO–HAp, and (c) 0.5 MgO–HAp. The insets along with yellow arrow show the selected facets of the SEM at which EDAX recorded. | |
 |
| | Fig. 6 Variation of (a) surface area vs. MgO content and (b) average pore diameter vs. MgO content. | |
3.7. Transmission electron microscopic analysis
Detailed transmission electron microscopy (TEM) (Fig. 7(a)–(d)) images revealed the presence of MgO (particle size ∼ 5 nm) and the 2D sheets with sufficient amount of porosity. HAp particles are represented by green arrows and dark places surrounded by yellow rings in the images 7(a)–(c) showed the MgO particles embedded in the HAp particles. Pores are indicated by red arrows in red colored rings. TEM results confirmed the crystalline HAp with variable porosity according to the MgO concentration. The selected area electron diffraction (SAED) pattern corresponding to the interplane distance (002) and (211) shown in Fig. 7(d), and depicts spotty ring patterns with the clear diffraction pattern and revealed the crystalline phase of HAp along with random orientation. This SAED analysis is consistent with XRD results which confirmed the formation of hexagonal porous structure of HAp. Porous structure of HAp is also of great interest to the orthopedic scientists. Porous HAp biomaterial interacts with natural tissues and the pores are providing scaffold to the bone tissue growth and regeneration well into the pores and also increases the strength of the implants. Porosity controls the mechanical as well as hydrophilic behavior of a material. Hydrophilic behavior of a material is strongly correlated with the porosity/density of the material. The mechanical properties (density, Young's modulus, fracture toughness, load capability etc.) of the prepared HAp biomaterials as studied earlier are already found to be strongly correlated to the porosity. As the porosity decreases, the density is increases which improve other mechanical properties of the material and vice versa. A reciprocal relationship of density, Young's modulus, and fracture toughness with porosity are obtained and these are highest at the lowest porosity (for 0.5 wt% MgO–HAp composite).
 |
| | Fig. 7 TEM images of nanostructures of porous HAp bioceramic composite (a) low magnification bright field image showing interconnected nanoparticles of MgO. (b) High-magnification bright field image showing irregular distribution of the porosity along with MgO nanoparticles. (c) TEM image showing uniform distribution of interconnected HAp sheets. (d) The selected area electron diffraction (SAED) pattern showing spotty hexagonal pattern of HAp. | |
3.8. Contact angle analysis
The surface reactivity of the prepared HAp and its composites with MgO has been analyzed by observing the contact angle between the surface of the material and distilled water. Hydrophilic characteristics of pure HAp and its composites are important to study the biocompatibility and degradability of the samples. Hydrophilic property of HAp and its composites controls the transfer of body fluid and cell suspension. Contact angle is the measure of hydrophilic behavior of a material, so contact angle for each of the sample, pure as well as composites has been measured and shown in Fig. 8(a)–(e). The measured values of the contact angles in degrees are listed in Table 1. Contact angle is significantly affected by MgO concentration. Table 1 also exhibits that the values of contact angle are found to be lowest (21.20°) for pure HAp and started to increase with increasing the wt% of MgO into HAp. It increased remarkably high (40.37°) for HAp composite with 5% of MgO content. Contact angle is achieved remarkably at peak value (40.37°) for 0.5 wt% of MgO–HAp composite having highest density and lowest porosity. Pure HAp is highly hydrophilic material and as the MgO is incorporated the reduction in hydrophilic nature is observed due to the lack of hydroxyl chain in MgO. The contact angle less than 40.37° for MgO–HAp indicate the hydrophilic nature of MgO–HAp samples for bioactive applications. The gradual increase in contact angle with increasing concentration of MgO attributed to decrease in hydrophilic nature and also decrease in the degradability of the material due to the formation of hydrophobic layer upon the surface of MgO–HAp composite samples. The thickness of this layer increases with increasing the MgO content. Thus the composite sample with highest concentration of MgO is the least degradable and most stable. The hydrophilic behavior and degradability of HAp–MgO material may be easily adjusted by varying the MgO concentration. MTT assay for cell viability for LE cells when treated with pure HAp and different wt% of MgO included HAp composites have been performed (Fig. 9). The above studies based on several mechanical parameters, cell viability and hydrophilic behavior of the prepared samples indicated the best composite having 0.5 wt% of MgO in HAp. Sintering temperature for best outcomes for each composition is also optimized and it is noticed to be 1200 °C, beyond this temperature the inverse effect of mechanical characteristics of pure as well as all the MgO–HAp composites is observed.
 |
| | Fig. 8 Figure shows the hydrophilic behavior of HAp and MgO–HAO samples measured by optical images of contact angle evaluated by using image J image analyzer software (a) pure HAp, (b) 0.1 MgO–HAp, (c) 0.2 MgO–HAp, (d) 0.3 MgO–HAp and (e) 0.5 MgO–HAp. | |
 |
| | Fig. 9 Cell viability after 72 h measured by MTT assay method using LE cells with different concentration of 0.5 MgO–HAp nanoparticles. | |
4. Conclusions
In this study, the pure HAp via microwave irradiation and MgO added HAp composites using ball milling method have been prepared successfully. The microwave irradiation technique reduces the secondary phase formation. Concentration of MgO into pure HAp and sintering temperature have been optimized on the several mechanical parameters viz. density, Young's modulus, fracture toughness, load capability, porosity and its hydrophilic nature. This study revealed that the HAp composite modified with 0.5 wt% of MgO in pure HAp and sintered at 1200 °C is extremely beneficial for densification to achieve the highest density, Young's modulus, fracture toughness and load capability. It is concluded that the mechanical properties, hydrophilic properties and degradability are strongly dependent on porosity. However, the porosity is easily controlled by MgO concentration as well as sintering temperature. The above composite sample exhibits highest mechanical properties and is promising for biomedical applications as suggested by MTT assay.
Acknowledgements
Dr C. R. Gautam highly acknowledges to University Grant Commission, New Delhi, India for providing the financial support under Raman Post-Doctoral Research Award Fellowship (Award No. F5-65/2014 (IC)). Authors would like to acknowledge to Prof. P. M. Ajayan and Robert Vajtai, Department of Materials Science and Nano Engineering, Rice University, Houston, Texas, USA for providing the necessary facilities required to carry out this research work.
References
- M. Niinomi, Metall. Mater. Trans. A, 2002, 33, 477–486 CrossRef.
- A. A. Oshkour, S. Pramanik, M. Mehrali, Y. H. Yat, F. Tarlochan and N. A. A. Osman, J. Mech. Behav. Biomed. Mater., 2015, 49, 321–331 CrossRef PubMed.
- D. A. Puleo and W. W. Huh, J. Appl. Biomater., 1995, 6, 109–116 CrossRef CAS PubMed.
- J. J. Jacobs, J. L. Gilbert and R. M. Urban, J. Bone Jt. Surg., 1998, 80, 268–282 CAS.
- J. Nagels, M. Stokdijk and P. M. Rozing, J. Shoulder Elbow Surg., 2003, 12, 35–39 CrossRef PubMed.
- T. G. Kim, S. Heungsoo and W. L. Dong, Adv. Funct. Mater., 2012, 22, 2446–2468 CrossRef CAS.
- H. L. Jang, K. Lee, C. S. Kang, H. K. Lee, H. Y. Ahn, H. Y. Jeong, S. Park, S. C. Kim, K. Jin, J. Park, T. Y. Yang, J. H. Kim, S. A. Shin, H. N. Han, K. H. Oh, Y. Lee, J. Jun Lim, K. S. Hong, M. L. Snead, J. Xu and K. T. Nam, ACS Nano, 2015, 9, 4447–4457 CrossRef CAS PubMed.
- K. Kandori, N. Horigami, A. Yasukawa and T. Ishikawa, J. Am. Ceram. Soc., 1997, 80, 1157–1164 CrossRef CAS.
- Li. Qian, Ma. Lie and G. Changyou, J. Mater. Chem. B, 2015, 3, 8921 RSC.
- C. M. Weaver, Curr. Osteoporos. Rep., 2014, 12, 211–218 CrossRef PubMed.
- G. Goller, H. Demirkiran, F. N. Oktar and E. Demirkesan, Ceram. Int., 2003, 29, 721–724 CrossRef CAS.
- R. Murugan and S. Ramakrishna, Compos. Sci. Technol., 2005, 65, 2385–2406 CrossRef CAS.
- A. W. Xu, Y. Ma and H. Colfen, J. Mater. Chem. B, 2007, 17, 415–449 RSC.
- G. Falini, J. Inorg. Mater., 2000, 2, 455–461 CrossRef CAS.
- T. Okuda, K. Ioku, I. Yonezawa, H. Minagi, Y. Gonda, G. Kawachi, M. Kamitakahara, Y. Shibata, H. Murayama, H. Kurosawa and T. Ikeda, Biomaterials, 2008, 29, 2719–2728 CrossRef CAS PubMed.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CAS.
- H. Petite, V. Viateau, W. Bensaid, A. Meunier, C. De Pollak and M. Bourguignon, Nat. Biotechnol., 2000, 18, 959–963 CrossRef CAS PubMed.
- S. Kehoe, J. Stokes, M. Ardhaoui and L. Looney, J. Mater. Process. Technol., 1999, 89–90, 373–377 Search PubMed.
- S. Fa-Nian, J. C. Almeida, L. A. Helguero, H. V. Maria Fernandes, C. Jonathan Knowles and J. Rocha, Inorg. Chem., 2015, 54, 9929–9935 CrossRef PubMed.
- M. Vallet-Regí, Dalton Trans., 2000, 97–108 Search PubMed.
- S. Bose, M. Roy and A. Bandyopadhyay, Trends Biotechnol., 2012, 30, 546–554 CrossRef CAS PubMed.
- D. Lickorish, L. Guan and J. E. Davies, Biomaterials, 2007, 28, 1495–1502 CrossRef CAS PubMed.
- R. K. Roeder, M. M. Sproul and C. H. Turner, J. Biomed.
Mater. Res., Part A, 2003, 67, 801–812 CrossRef PubMed.
- A. Bigi, G. Cojazzi, S. Panzavolta, A. Ripamonti, N. Roveri, M. Romanello, K. Noris Suarez and L. Moro, J. Inorg. Biochem., 1997, 68, 45–51 CrossRef CAS PubMed.
- W. Suchanek and M. Yoshimura, J. Mater. Sci., 1998, 13, 94–117 CAS.
- Z. Yazdanpanah, M. E. Barololoom and B. Hashemi, J. Mech. Behav. Biomed. Mater., 2015, 41, 36–42 CrossRef CAS PubMed.
- M. Español, G. Mestres, T. Luxbacher, J. B. Dory and M. P. Ginebra, ACS Appl. Mater. Interfaces, 2016, 8, 908–917 Search PubMed.
- J. J. Li, L. David Kaplanb and H. Zreiqat, J. Mater. Chem. B, 2014, 2, 7272–7306 RSC.
- O. Hiromichi, M. Yu, Y. Shin, A. Nobuo and O. Mitsuo, J. Biomed. Mater. Res., Part A, 2006, 79A, 329–337 CrossRef PubMed.
- Y. L. Hong, H. S. Fan, B. Li, B. Guo, M. Liu and X. D. Zhang, Mater. Sci. Eng., R, 2010, 70, 225–242 CrossRef.
- L. J. Jha, J. C. Best, L. Knowles, I. Rehman, J. D. Santos and W. Bonfield, J. Mater. Sci.: Mater. Electron., 1997, 8, 185 CrossRef CAS.
- J. Li, L. Hermansson and R. Soremark, J. Mater. Sci.: Mater. Med., 1993, 4, 50–54 CrossRef CAS.
- R. Tolouei, C. Y. Tan, S. Ramesh, I. Sopyan and W. D. Teng, Adv. Mater. Res., 2011, 264, 1832–1838 CrossRef.
- S. Heinemann, C. Heinemann, M. J. Eager, J. Neunzehn, H. P. Wiesmann and T. Hanke, ACS Appl. Mater. Interfaces, 2011, 3, 4323–4331 CAS.
- W. J. E. M. Habraken, L. T. De Jonge, J. G. C. Wolke, L. Yubao, A. G. Mikos and J. A. Jansen, Adv. Drug Delivery Rev., 2007, 59, 234–248 CrossRef CAS PubMed.
- J. A. Delgado, L. Morejon, S. Martınez, M. P. Ginebra, N. Carlsson, E. Fernandez, J. A. Planell, M. T. Clavaguera-Mora and J. Rodriaguez-Viejo, J. Mater. Sci.: Mater. Med., 1999, 10, 715–719 CrossRef CAS PubMed.
- C. R. Gautam, M. Tamuk, C. W. Manpoong, S. S. Gautam, S. Kumar, A. K. Singh and V. K. Mishra, J. Mater. Sci., 2016, 51, 4973–4983 CrossRef CAS.
- M. K. Young, S. Kim and H. E. Kim, J. Am. Ceram. Soc., 1999, 82, 2963–2968 Search PubMed.
- M. Mehrali, E. Moghaddam, S. F. Shirazi, S. Baradaran, M. Mehrali, S. T. Latibari, H. S. Metselaar, N. A. Kadri, K. Zandi and N. A. Osman, ACS Appl. Mater. Interfaces, 2014, 6, 3947–3962 CAS.
- O. Gunduz, C. Gode, Z. Ahmad, H. Gokce, M. Yetmez, C. Kalkandelen, Y. M. Sahin and F. N. Oktar, J. Mech. Behav. Biomed. Mater., 2014, 35, 70–76 CrossRef CAS PubMed.
- S. Ramesh, C. Y. Tan, C. L. Peralta and W. D. Teng, Sci. Technol. Adv. Mater., 2007, 8, 257–263 CrossRef CAS.
- M. Aminzare, A. Eskandari, M. H. Baroonian, A. Berenov, Z. R. Hesabi, M. Taheri and S. K. Sadrnezhaad, Ceram. Int., 2013, 39, 2197–2206 CrossRef CAS.
- M. Schumacher and M. Gelinsky, J. Mater. Chem. B, 2015, 3, 4626–4640 RSC.
- J. Li, L. Cao, Y. Song, S. Zhang, C. Zhao, F. Zhang and X. Zhang, Bioinorg. Chem. Appl., 2011, 2011, 1–7 CrossRef PubMed.
- M. P. Staiger, A. M. Pietak, J. Huadmai and G. Dias, Biomaterials, 2006, 27, 1728–1734 CrossRef CAS PubMed.
- W. Cui, E. Beniash, E. Gawalt, Z. Xu and C. Sfeir, Acta Biomater., 2013, 98, 650–8659 Search PubMed.
- X. Gu, Y. Cheng, Y. Zheng, S. Zhong and T. Xi, Biomaterials, 2009, 30, 484–498 CrossRef CAS PubMed.
- Le. Ferrand, H. Bouville, F. T. P. Niebel and A. R. Studart, Nat. Mater., 2015, 14, 1172–1179 CrossRef PubMed.
- L. Bertinetti, A. Tampieri, E. Landi, G. Martra and S. Coluccia, J. Eur. Ceram. Soc., 2006, 26, 987–991 CrossRef CAS.
- T. J. Webster, E. A. Massa-Schlueter, J. L. Smith and E. B. Slamovich, Biomaterials, 2004, 25, 2111–2121 CrossRef CAS PubMed.
- V. K. Mishra, B. N. Bhattacharjee, O. Parkash, D. Kumar and S. B. Rai, J. Alloys Compd., 2014, 614, 283–288 CrossRef CAS.
- R. Tolouei, S. Ramesh, C. Y. Tan, M. Amiriyan, L. Samuel and B. K. Yap, Solid State Sci. Technol., 2011, 19, 137–143 CAS.
- S. Srinivasan, H. Gareth McKinley and E. Robert Cohen, Langmuir, 2011, 27, 13582–13589 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.