
An ultrasound-assisted ion association dispersive liquid–liquid microextraction coupled with micro-volume spectrofluorimetry for chromium speciation
Waqas Ahmad,
A. Al-Sibaai,
A. S. Bashammakh,
H. Alwael and
M. S. El-Shahawi†
*
Department of Chemistry, Faculty of Science, King Abdulaziz University, P. O. Box 80203, Jeddah 21589, Kingdom of Saudi Arabia. E-mail: malsaeed@kau.edu.sa; mohammad_el_shahawi@yahoo.co.uk; Fax: +966-12-6952292; Tel: +966-12-6952000 ext. 64422
First published on 8th July 2016
Abstract
A micro-volume spectrofluorimetric coupled ultrasound-assisted ion association dispersive liquid–liquid microextraction (USA-IA-DLLME) procedure for the total determination and speciation of chromium(III & VI) species has been established. This demonstration was based upon the formation of a complex ion associate between halochromate (CrO3Cl−) and rhodamine 6G hydrochloride dye (RG+). At optimal conditions, the method exhibited a three order of magnitude wide linear concentration range (1.0–1000 μg L−1), with detection and quantification limits of 0.57 μg L−1 and 1.9 μg L−1, respectively. Competent cations, anions and oxoanions did not interfere with the chromium(VI) determination. Chromium(III) was also evaluated after its conversion to chromate with H2O2 in alkaline media. The nature of the extractant and the disperser, their volumes and the extraction time were optimized. The fluorescence quenching mechanism of the complex ion associate was discussed. The method was validated by the determination of chromium(VI) in real water (sea and tap water) samples. The results were also compared successfully with those from inductively coupled plasma-optical emission spectrometry (ICP-OES) in terms of the student’s t- and F test data at 95% confidence.
1. Introduction
Microextraction techniques, in particular dispersive liquid–liquid microextraction (DLLME), are infrequently coupled to electrochemical techniques and fluorimetry.1 The challenges in developing ion pairing fluorescent reagents for various analytes and higher extraction volumes make microextraction techniques less convenient for fluorimetry, a well established science.1 According to the Scopus database, every year approximately three hundred studies report dispersive liquid–liquid microextraction (DLLME) for a wide range of analytes. It is mostly coupled with chromatographic techniques (GC, HPLC) and spectroscopy (AAS, AES & spectrophotometry).1,2 The percentage of reports of DLLME coupled with spectrofluorimetric methods is negligible as compared to its coupling with GC & HPLC.1–3 Fluorescence spectrometry is a well established quantitative tool for analysis. It possesses high sensitivity, selectivity, low cost and simplicity compared to other techniques requiring heavy instrumentation and/or tedious processes.DLLME has been used for the analysis of various analytes including chromium(VI). Its hexavalent state is considered a potential carcinogen affecting genetic makeup and damaging DNA. Chronic exposure and skin exposure lead to pulmonary damage and dermatitis.4 The permissible range set by WHO and US-EPA for chromium(VI) in drinking water equals 0.05 and 0.1 μg mL−1, respectively.5 Several analytical methods are known for chromium determination including atomic absorption spectrometry (AAS),5–18 HPLC,19–24 inductively coupled plasma mass spectrometry (ICP-MS),25,26 gas chromatography (GC),27 spectrofluorimetry,28–32 spectrophotometry,33–38 and Atomic emission spectrometry (AES).39
These methods (GC, HPLC, ICP-OES/MS, etc.) are the most reliable even though they possess numerous shortcomings. Some require heavy and expensive instrumentation, consuming toxic and costly organic solvents in large quantities, while others are slow and experience interference and sensitivity issues. Moreover, in several cases the total determination of chromium is ignored. Among these techniques, spectrofluorimetry has a unique dominance and lead in sensitivity, selectivity, low cost and simplicity. Its combination with DLLME will be highly convenient and would be considered a routine method for any analyte. Thus, a comprehensive and green analytical preconcentration method coupled with spectrofluorimetry for chromium was very much needed.
Herein, the first inceptive ultrasound assisted ion association dispersive liquid–liquid microextraction (USA-IA-DLLME) combined with micro-volume spectrofluorimetry for chromium(III & VI) speciation is reported. Rhodamine 6G chloride (RG+), a fluorescent dye,40 was employed for the selective extraction of the metal ion. The dye serves two functions. Firstly, it acts as an ion pairing reagent in the preconcentration step and secondly, as a fluorescent probe in sensing. This study covers and discusses (i) the optimization of USA-IA-DLLME parameters, (ii) the detailed fluorescence quenching mechanism and calculations of parameters like quenching and binding constants, (iii) chromium speciation, (iv) analytical applications and validation studies by various statistical tests, (v) the impact of diverse ions, (vi) the characterization of complex ion associates, and (vii) future trends.
2. Experimental
2.1. Reagents and materials
All chemicals and solvents utilized were of analytical reagent grade. A stock solution of 1000 μg mL−1 was prepared using K2Cr2O7 from BDH chemicals Ltd, Poole UK. Working solutions were made by adding the appropriate volumes from the stock solution. Solutions of interfering metals ions are made from their chloride and nitrate salts. A 0.1% m/v stock solution of RG+ (Sigma-Aldrich) was prepared by accurately dissolving the appropriate weight in deionized water. A BDH hydrogen peroxide, H2O2 (30% v/v), was also used as an oxidizing agent for the oxidation of chromium(III) in an aqueous alkaline solution of KOH (0.2 mol L−1). A series of Britton–Robinson (B–R) buffers (pH 2–11.4) were prepared in deionized water. BDH mineral acids (HCl, HNO3, H2SO4, H3PO4, HClO4) were used.2.2. Apparatus
A Perkin-Elmer (USA) LS55 spectrofluorimeter with a 10 mm quartz microcuvette (100 μL) was used for the fluorescence measurements. As a reference method inductively coupled plasma optical emission spectrometry (Perkin Elmer, California, CT USA) was utilized for chromium determination. A digital micropipette (model H100) and pH meter (inoLab pH/ion level 2) were used for the dilution of the chromium(VI) solution and pH measurements, respectively. Ultra pure water was obtained from a Milli-Q Plus system from Millipore; Bedford, MA, USA.2.3. Recommended procedures
| ΔI (%) = (I0 − If)/I0 × 100 | (1) |
3. Results and discussion
In the present work an USA-IA-DLLME coupled micro-volume spectrofluorimetric method for the determination of chromium in real water samples is proposed. It is based on the extraction of chromium(VI) and RG+ as a complex ion associate [RG+·CrO3Cl−] into chloroform in acidic media. The free reagent is extracted into chloroform and shows intense fluorescence at λex/em = 525/570 nm; its fluorescence intensity is quenched in the presence of chromium(VI) in acidic media with minor wavelength shifts confirming the formation of a complex ion associate [CrO3Cl−·RG+] (Fig. 1 and 2). This approach of associating the decrease in fluorescence to the chromium(VI) concentration was employed in spectrofluorimetry coupled USA-IA-DLLME. Microextraction techniques, mainly DLLME coupled spectrofluorimetry, have never been reported for hexavalent chromium determination. The reason may be the incompatibility with small extraction solvent volumes and the non-availability of ion pairing fluorescent probes. A suitable ultra microcuvette, RG+ as a fluorescent sensor and as an ion-pairing reagent makes spectrofluorimetry coupled DLLME highly convenient for chromium speciation.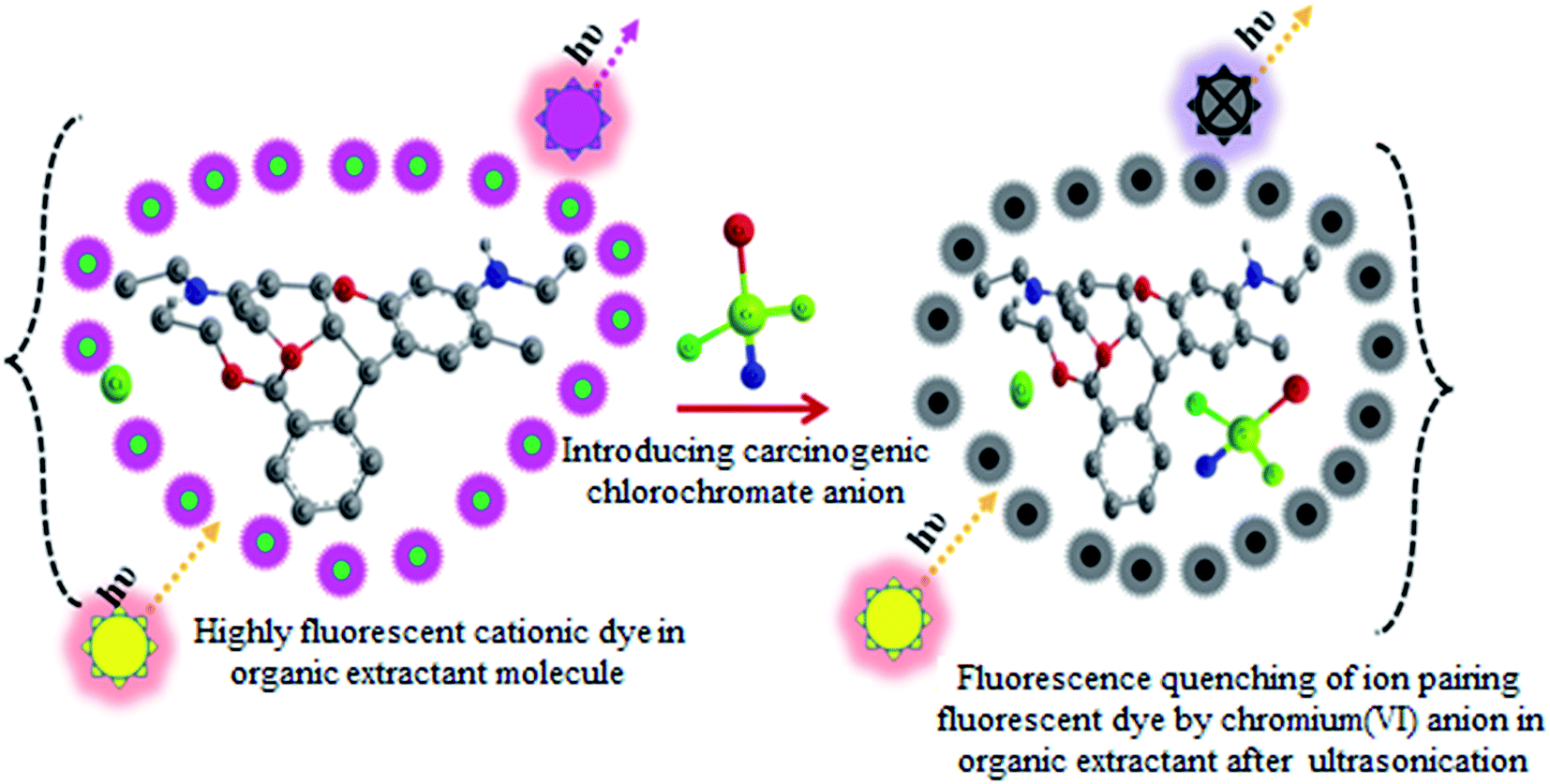 | ||
| Fig. 1 A schematic illustration of the sensing principle for carcinogenic chromium(VI) detection based on the fluorescence quenching of a cationic laser dye in acidic media into an organic extractant in USA-IA-DLLME. | ||
 | ||
| Fig. 2 Fluorescence spectra of rhodamine 6G, 1.0 × 10−5 mol L−1 (1) and the complex ion associate of chromium(VI), 300 μg L−1 (2) in 0.5 M HCl and pH < 1.0 at 570 nm. | ||
3.1. Optimization of analytical parameters
Acidity contributes substantially to any method development. The influence of various mineral acids like hydrochloric, sulphuric, nitric and perchloric acids on the extraction of chromium reveals that HCl is the most suitable mineral acid. The influence of mineral acids on the extraction percentage follows the order HCl > HNO3 > H2SO4 > HClO4.The selection of HCl is followed by its concentration optimization. A concentration of 0.5 mol L−1 gave maximum quenching. At higher acid concentrations ΔI decreased possibly because of the reduction of chromium(VI) to other non-extractable forms (Fig. 3a).
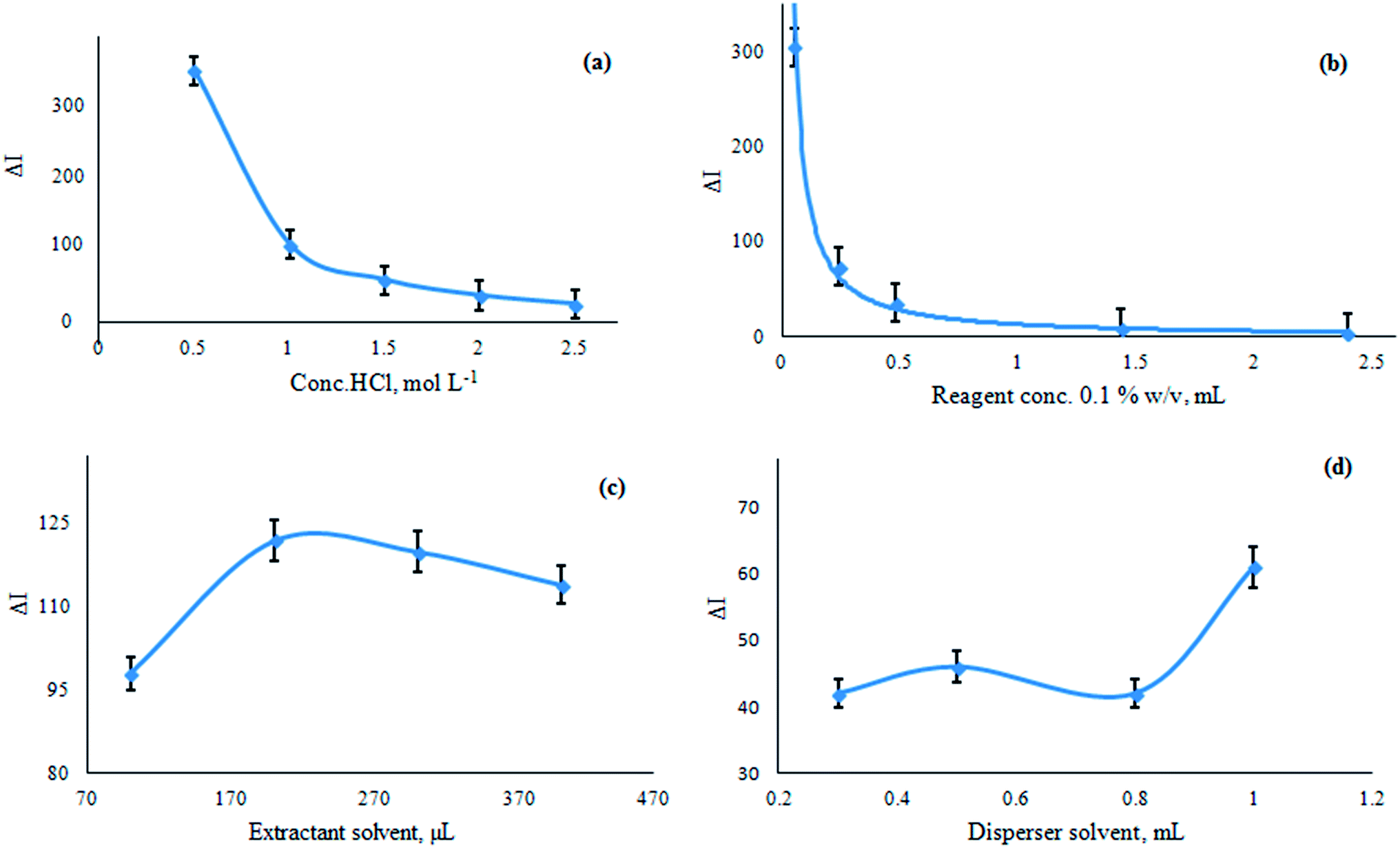 | ||
| Fig. 3 The influence of various parameters: (a) HCl concentration, 0.5–2.5 mol L−1 (b) RG+ concentration, 0.3–1.0 mL, (c) extractant solvent volume (100–400 μL), (d) disperser solvent volume (0.3–1.0 mL). | ||
The most predominant species at 0.5 mol L−1 (pH < 1.0) is the hydrogen chromate oxoanion (HCrO4−).41 In HCl media (0.5 mol L−1), it is converted to the chlorochromate anion (CrO3Cl−) via the following equation:42
| HCrO4− + H+ + Cl− ⇌ CrO3Cl− + H2O | (2) |
In aqueous HCl media, the resultant CrO3Cl− species are directly proportional to the formation and extraction of the complex ion associate in the organic phase according to the following equations:
| CrO3Claq− + RGaq+ ⇌ [RG+·CrO3Cl−]aq | (3) |
| [RG+·CrO3Cl−]aq + nSorg ⇌ [RG+·CrO3Cl− nS]org | (4) |
The selection of the organic solvent is a very critical step in any method development. Several organic solvents like carbon tetrachloride, chloroform, dichloromethane, cyclohexane, toluene, benzene, and n-hexane have been tested as extraction solvents. The most effective solvent was chloroform for the extraction of the complex ion associate (RG+·CrO3Cl−).
The concentration of the reagent (RG+) was optimized (Fig. 3b). The change in fluorescence intensity (ΔI) increased up to 0.025 mL (0.1% w/v) RG+, whereas increasing the concentration results in lowering the fluorescence, becoming almost negligible at higher concentrations. Therefore, 1.0 × 10−5 mol L−1 was adopted as the optimum.
The effect of the shaking time on the fluorescence intensity of the complex ion associate was investigated to check the reaction time at different time intervals (0.5–4.0 min). In DLLME, the injections of the blend of extractant and disperser solvents causes the formation of tiny droplets of extraction solvents with infinite surface area for the analyte, hence prompt mass transfer occurs, making the extraction time insignificant. Lastly, varying the salt concentration (0.0–15% w/v) revealed no significant change in extraction performance.
3.2. Selectivity
The selectivity and reliability of the proposed procedure was inspected in the presence of relatively high concentrations (50–500 μg L−1) of various interfering ions. The tolerance limit (w/w) for the interfering ion corresponds to a relative error within ±5% from the true value for the developed complex ion associate [RG+·CrO3Cl−]. The results reveal (Table 1) no interference from an excess concentration of a diverse range of cations and anions. The tolerance limit for most common interfering anions like IO4−, IO3− was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 without the addition of any masking agent. The interference of MnO4− and WO4− was masked by adding a few drops of NaN3 (0.1% w/v) and thiocyanate ions (0.01% m/v), respectively. The addition of a few drops of NaN3 (0.1% w/v) and CNS− ions reduces MnO4− and WO4− to some un-extractable species of Mn2+ and tungsten species with oxidation states between III and V,43 respectively.
:100 without the addition of any masking agent. The interference of MnO4− and WO4− was masked by adding a few drops of NaN3 (0.1% w/v) and thiocyanate ions (0.01% m/v), respectively. The addition of a few drops of NaN3 (0.1% w/v) and CNS− ions reduces MnO4− and WO4− to some un-extractable species of Mn2+ and tungsten species with oxidation states between III and V,43 respectively.
| Interference species | Interferent-to-analyte ratio |
|---|---|
| Li+, Ca2+, Al3+, Zn2+, Mg2+, Cd2+, Ba2+, Co2+, Cr3+, Hg2+, Cu2+, Pb2+, F−, Br−, CH3COO−, NH4+, Sn2+,IO4−, IO3− | 1:100 |
| MnO4−, WO42− | 1:5 |
3.3. Investigation of the USA-IA-DLLME procedure
DLLME parameters were optimized by performing various trial experiments and their impact on the fluorescence of the sedimented phase was assessed. In USA-IA-DLLME, the fast injection of the mixture of chloroform and methanol as extractant and disperser solvents results in turbidity followed by formation of very nice minute droplets. The droplets of extractant were dispersed by ultrasonication. The sedimented organic phase was introduced into a microquartz cell (100.0 μL) for fluorescence measurements at λex/em = 525/570 nm after centrifugation. The parameters of USA-IA-DLLME like the nature and volume of the extractant and disperser solvents, along with the extraction time were optimized.The following series of solvents was tested: carbon tetrachloride, tetrachloride, n-hexane, dichloromethane, cyclohexane, chlorobenzene, ethyl acetate, chloroform toluene and benzene. Chloroform extracts the free reagent as well as the complex ion associate whereas toluene and benzene extract only the complex ion associate. However, chloroform was more appropriate in spectrofluorimetric determination and its higher density makes it more suitable for DLLME. Moreover, it offers less instrumental error and provides good emission spectra. The influence of the extraction solvent volume was investigated by varying the volume (100–400 μL) as depicted in Fig. 3c The volume of the sedimented phase and the fluorescence signal were less when the volume of the extraction solvent was between 100 and 150 μL. The maximum fluorescence intensity and a higher sedimented phase volume was seen when the extraction solvent volume was 200 μL. At higher extractant volumes, the amount of sedimented phase increases but the fluorescence intensity decreases. Therefore, 200 μL chloroform was chosen as the optimum volume.
The stability and equilibrium of the ternary phase system can be altered by various disperser solvents like methanol, ethanol, acetone, acetonitrile, tetrahydrofuran, etc. Acetone, acetonitrile and methanol were tested. Methanol was selected as the value of ΔF was higher compared to the others upon its combination with chloroform. Moreover the combination resulted in very fine visible droplets as compared to the other dispersers. The selection of the disperser is equally as important as the extraction solvent. Mostly, lower disperser volumes result in an unsatisfactory partition of the extractant as well ineffective droplet formation, ultimately decreasing the extraction efficiency. The solubility of the analyte increases at higher disperser volumes reducing the polarity of the aqueous phase, and ultimately the analyte distribution moves down towards the extraction solvent affecting the extraction efficiency. If the solubility is much higher in the extraction solvent, it will be sedimented along with the organic phase, thus reducing the extraction efficiency. Various volumes of methanol (0.3–1.0 mL) and chloroform (200 μL) were assayed. The results in Fig. 3d show that 1.0 mL of methanol gives a maximum and constant signal and thus, this volume was selected as the optimum.
The large surface area available for the transformation of the analyte makes the extraction time insignificant because of the prompt extraction. However, the extraction time and its effect on the extraction over a period of 1.0–5.0 min were studied. The equilibrium state was established promptly revealing its insignificance. Therefore, a one minute extraction time was adopted as the optimum.
3.4. Fluorescence quenching mechanism
Different kinds of fluorescence quenching mechanism are known.32,44 The most common quenching process includes (i) excited-state complex formation, (ii) an energy transfer inter-system crossing, (iii) singlet-to-triplet excitation and finally (iv) an excited inter-system crossing. Thus, the fluorescence quenching of RG+ using chromium(VI) as a quencher was critically studied at room temperature using the steady state method.44 The fluorescence quenching of RG+ (λmaxex/λmaxem 525/570 nm) in the presence of different concentrations (0.5–6.0 μg mL−1) of chromium(VI) as a quencher is shown in Fig. 4. The corresponding fluorescence intensity (F) against the reagent blank at the optimum conditions was finally measured. The emission power of RG+ decreases at regular intervals with an increase in the quencher concentration as is evident from the Stern–Volmer model. The Stern–Volmer constant was calculated using equation:45| I0/I = 1 + Ksv[Q] | (5) |
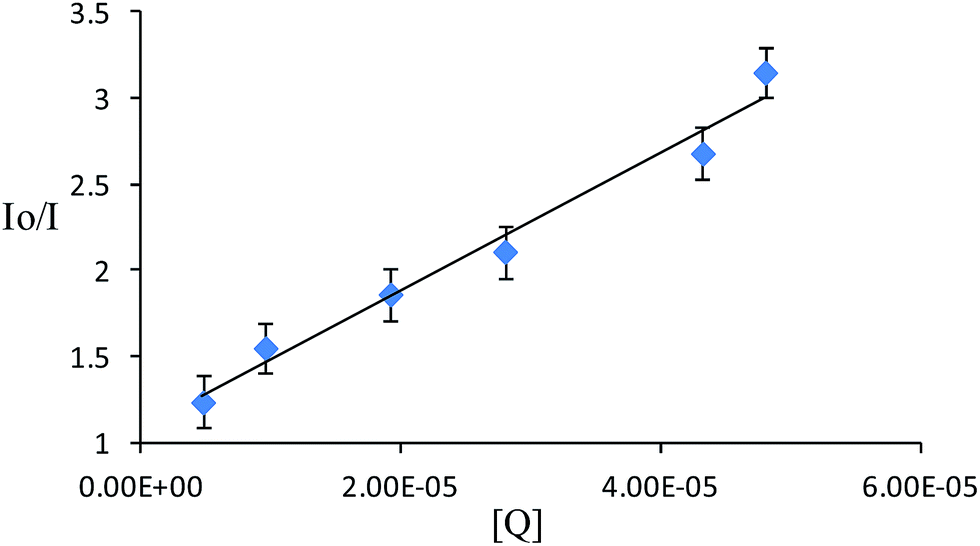 | ||
| Fig. 4 Stern–Volmer plot for the fluorescence quenching of RG+ by Cr(VI). | ||
I0 and I are the signal intensities before and after the addition of halochromate, respectively. Ksv is the Stern–Volmer quenching constant and [Q] is the concentration of the quencher. The Stern–Volmer plot for RG+ is shown in Fig. 4. The calculated Ksv and correlation factor were found to equal 4.01 × 104 L mol−1 and 0.978, respectively.
The apparent binding constant (KA) and the number of binding sites (n) were computed by employing the Benesi–Hildebrand fluorescence model46
|
log(I0/I)/I = logKA + nlog[Q]
| (6) |
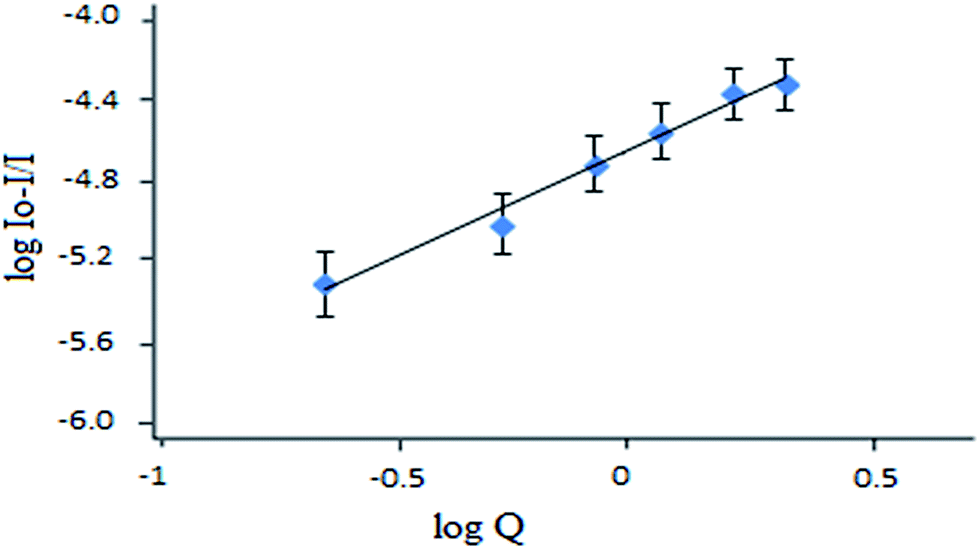 | ||
| Fig. 5 The Benesi–Hildebrand plot for the calculation of the binding constant for chromium(VI) with the reagent RG+. | ||
3.5. Analytical figure of merit
The repeatability, linearity, and detection and quantification limits were determined at optimized conditions. A linear plot at λex/em = 525/570 nm was achieved by plotting the quenched fluorescence intensity of the complex ion associate [RG+·CrO3Cl−] versus chromium(VI) concentrations. The plot was linear over the range of 100–1000 μg mL−1. The correlation coefficient (R2) was 0.9892 and regression equation was:| y = 0.1034C (μg mL−1) + 24.028. | (7) |
The lower limits of detection (LOD) and quantification (LOQ) obtained according to IUPAC47 and were 0.57 μg L−1 and 1.9 μg L−1, respectively. The figures of merit for some reported spectrofluorimetric methods for chromium determination were compared28–32 and the results are summarized in Table 2. Several methods among the reported show serious interference by halide ions and in some cases a high LOD, whereas the developed method offers a better LOD, ease of use and less interference. The LOD of the method is lower than the maximum allowable level of chromium reported by WHO in soil, solid sewage and water.
| λex/em (nm) | Preconcentration method | Linear range | LOD (μg L−1) | Remarks | Ref. |
|---|---|---|---|---|---|
| 525/545 | In aqueous media | 8–80 ng mL−1 | 0.8 μg L−1 | No speciation, interference due to Hg(II), Pb(II), Fe(III), Cu(II) | 31 |
| 540/560 | In aqueous media | 0.01–0.20 μg mL−1 | 3 μg L−1 | No speciation, lower sensitivity | 32 |
| 530/550 | In aqueous media (Tween 80) | 2.0–100.0 ng mL−1 | 0.37 μg L−1 | BrO3−, IO3−, MnO4− and NO2− interfere seriously | 33 |
| 295/345 | In aqueous media | 1.0 × 10−7 to 6.0 × 10−5 mol L−1 | 0.54 μg L−1 | No speciation | 34 |
| 242/305 | LLE | 1–114 μg L−1 | 0.43 μg L−1 | Sensitive, selective, higher volumes of organic solvent | 35 |
| 525/570 | DLLME | 1–1000 μg L−1 | 0.57 μg L−1 | Highly sensitive, selective and fast, green analytical method | This work |
3.6. Analytical applications
| Added (μg mL−1) | Founda (μg mL−1) | Recoveredb (%) | |||
|---|---|---|---|---|---|
| Cr(III) | Cr(VI) | Cr(III) | Cr(VI) | Cr(III) | Cr(VI) |
| a Average of three measurements ± standard deviation.b Average of the recovery ± relative standard deviation. | |||||
| 0.25 | 0.25 | 0.246 ± 0.004 | 0.243 ± 0.004 | 98.4 ± 1.44 | 97.3 ± 0.49 |
| 0.5 | 0.5 | 0.481 ± 0.011 | 0.475 ± 0.019 | 96.2 ± 1.94 | 95.06 ± 3.1 |
| Sample | Chromium(VI) added (μg L−1) | Chromium(VI) founda (μg L−1) | Recoveredb (%) | ||
|---|---|---|---|---|---|
| A | B | A | B | ||
| a Average of five measurements ± standard deviation.b Average of the recovery ± relative standard deviation.c The figures in parentheses are the corresponding theoretical values at P = 0.05. | |||||
| Tap water | 0.5 | 0.532 ± 0.02 | 0.51 ± 0.025 | 106.33 ± 3.8 | 102.6 ± 4.1 |
| t = 0.39 (2.78)c | |||||
| F = 1.56 (6.39) | |||||
| Tap water | 0.75 | 0.779 ± 0.19 | 0.7363 ± 0.02 | 102.3 ± 2.58 | 100.04 ± 2.85 |
| t = 0.072 (2.78) | |||||
| F = 1.07 (6.39) | |||||
| Sea water | 0.5 | 0.522 ± 0.025 | 0.545 ± 0.028 | 104.6 ± 5.19 | 109 ± 5.71 |
| t = 0.45 (2.78) | |||||
| F = 1.25 (6.39) | |||||
| Sea water | 0.75 | 0.783 ± 0.012 | 0.81 ± 0.016 | 103.93 ± 1.63 | 107.6 ± 2.05 |
| t = 0.13 (2.78) | |||||
| F = 1.77 (6.39) | |||||
4. Conclusion and future trends
The current study reports the first inception of DLLME coupled spectrofluorimetry for chromium determination. To sum up, a fast, simple and eco-friendly USA-IA-DLLME procedure for the total speciation and determination of chromium(III,VI) is presented. This scientific finding represents an advancement in microextraction methodology, assembling extensive studies on the technique as well as comprehensive discussion on the mechanism of action unlike the majority of DLLME literature. Comparisons in Table 2 highlight the clear significance over the literature studies. The method exhibits high selectivity towards strongly interfering oxoions in addition to common ions relevant to water. The developed technique represents a powerful aid in accelerating the extraction and separation processes e.g. the integration and mass transfer between various immiscible phases. A re-emergence of this approach may therefore bring novel opportunities for the development of sustainable analytical methodologies and the trend will continue in the future despite challenges faced for developing ion pairing reagents and fluorescent probes for sensing various analytes. The current work can be upgraded to picomolar levels via online preconcentration through nanosized packed solid phase extractor columns. Lastly, the present work will set the trend for coupling spectrofluorimetry with microextraction techniques and will serve as a standard and direction for researchers in the field.References
- W. Ahmad, A. A. Al-Sibaai, A. S. Bashammakh, H. Alwael and M. S. El-Shahawi, Recent advances in dispersive liquid–liquid microextraction for pesticide analysis, Trends. Anal. Chem., 2015, 72, 181–192 CrossRef CAS.
- H. Yahyavi, M. Kaykhaii and M. Hashemi, A rapid spectrofluorimetric method for the determination of malondialdehyde in human plasma after its derivatization with thiobarbituric acid and vortex assisted liquid–liquid microextraction, RSC Adv., 2016, 6, 2361–2367 RSC.
- M. Kaykhaii, K. H. Yahyavi, M. Hashemi and M. R. Khoshroo, A simple graphene-based pipette tip solid-phase extraction of malondialdehyde from human plasma and its determination by spectrofluorometry, Anal. Bioanal. Chem., 2016, 1–9 Search PubMed.
- A. Sunil and S. Jagadeeswara Rao, Photometric and fluorimetric determination of chromium(VI) using metal-oxo mediated reaction of 1-(2-hydroxyphenyl) thiourea in micellar medium, J. Anal. Chem., 2015, 70(2), 159–165 CrossRef CAS.
- S. Sadeghi and A. Z. Moghaddam, Multiple response optimization of sequential speciation of chromium in water samples by in situ solvent formation dispersive liquid–liquid microextraction prior to electrothermal atomic absorption spectrometry determination, J. Iran. Chem. Soc., 2015, 13, 117–130 CrossRef.
- A. S. Silva, G. C. Brandao, G. D. Matos and S. L. Ferreira, Direct determination of chromium in infant formulas employing high-resolution continuum source electrothermal atomic absorption spectrometry and solid sample analysis, Talanta, 2015, 144, 39–43 CrossRef CAS PubMed.
- M. Aghamohammadi, M. Faraji, P. Shahdousti, H. Kalhor and A. Saleh, Trace determination of lead, chromium and cadmium in herbal medicines using ultrasound-assisted emulsification microextraction combined with graphite furnace atomic absorption spectrometry, Phytochem. Anal., 2015, 26, 209–214 CrossRef CAS PubMed.
- I. López-García, Y. Vicente-Martínez and M. Hernández-Córdoba, Non-chromatographic speciation of chromium at sub-ppb levels using cloud point extraction in the presence of unmodified silver nanoparticles, Talanta, 2015, 132, 23–28 CrossRef PubMed.
- W. Xiong, C. Cheng and Y. Yang, Determination of total chromium in tea samples by suspension dispersive solid phase extraction combined with silver nanoparticles and using flame atomic absorption spectrometry, Anal. Methods, 2015, 7, 2093–2099 RSC.
- B. Leśniewska, L. Trzonkowska, E. Zambrzycka and B. Godlewska-Żyłkiewicz, Multi-commutation flow system with on-line solid phase extraction exploiting the ion-imprinted polymer and FAAS detection for chromium speciation analysis in sewage samples, Anal. Methods, 2015, 7, 1517–1526 RSC.
- A. Béni, R. Karosi and J. Posta, Speciation of hexavalent chromium in waters by liquid–liquid extraction and GFAAS determination, Microchem. J., 2007, 85, 103–108 CrossRef.
- I. López-García, M. Briceño, Y. Vicente-Martínez and M. Hernández-Córdoba, Ultrasound-assisted dispersive liquid–liquid microextraction for the speciation of traces of chromium using electrothermal atomic absorption spectrometry, Talanta, 2013, 11, 166–171 CrossRef PubMed.
- N. N. Meeravali, R. Manjusha and S. J. Kumar, A novel non-chromatographic strategy for the sequential/simultaneous extraction and analysis of chromium species by electrothermal atomic absorption spectrometry in effluents and different water sources, J. Anal. At. Spectrom., 2014, 29, 2168–2175 RSC.
- R. P. Monasterio, J. C. Altamirano, L. D. Martínez and R. G. Wuilloud, A novel fiber-packed column for on-line preconcentration and speciation analysis of chromium in drinking water with flame atomic absorption spectrometry, Talanta, 2009, 77, 1290–1294 CrossRef CAS PubMed.
- P. Hemmatkhah, A. Bidari, S. Jafarvand, M. R. M. Hosseini and Y. Assadi, Speciation of chromium in water samples using dispersive liquid–liquid microextraction and flame atomic absorption spectrometry, Microchim. Acta, 2009, 166, 69–75 CrossRef CAS.
- P. C. Pereira Lara, J. Nicácio Silveira, W. Borges Neto, M. A. Beinner and J. B. da Silva, Use of multivariate optimization to develop a method for direct chromium determination in breast milk by GF-AAS using aqueous calibration, J. chem. pharm. Res., 2015, 7, 1900–1906 Search PubMed.
- A. R. Borges, L. L. François, E. M. Becker, M. G. R. Vale and B. Welz, Method development for the determination of chromium and thallium in fertilizer samples using graphite furnace atomic absorption spectrometry and direct solid sample analysis, Microchem. J., 2015, 119, 169–175 CrossRef CAS.
- Z. TaoHong, L. Jin, P. QingZhi, Z. Ying and L. Jie, Direct determination of trace chromium in milk powder by Graphite Furnace Atomic Absorption Spectrometry with suspension sampling, J. Food Saf. Food Qual., 2015, 6, 1478–1482 Search PubMed.
- L. L. Wang, J. Q. Wang, Z. X. Zheng and P. Xiao, Cloud point extraction combined with high-performance liquid chromatography for speciation of chromium(III) and chromium(VI) in environmental sediment samples, J. Hazard. Mater., 2010, 177, 114–118 CrossRef CAS PubMed.
- A. N. Tang, D. Q. Jiang, Y. Jiang, S. W. Wang and X. P. Yan, Cloud point extraction for high-performance liquid chromatographic speciation of Cr(III) and Cr(VI) in aqueous solutions, J. Chromatogr. A, 2004, 1036, 183–188 CrossRef CAS PubMed.
- J. Sun, Z. Yang, H. Lee and L. Wang, Simultaneous speciation and determination of arsenic, chromium and cadmium in water samples by high performance liquid chromatography with inductively coupled plasma mass spectrometry, Anal. Methods, 2015, 7, 2653–2658 RSC.
- S. Catalani, J. Fostinelli, M. E. Gilberti and P. Apostoli, Application of a metal free high performance liquid chromatography with inductively coupled plasma mass spectrometry (HPLC-ICP-MS) for the determination of chromium species in drinking and tap water, Int. J. Mass spectrom., 2015, 387, 31–37 CrossRef CAS.
- P. Li, L. M. Li, J. Xia, S. Cao, X. Hu, H. Z. Lian and S. Ji, Determination of hexavalent chromium in traditional Chinese medicines by high performance liquid chromatography with inductively coupled plasma mass spectrometry, J. Sep. Sci., 2015, 38(23), 4043–4047 CrossRef CAS PubMed.
- V. Vacchina, I. de la Calle and F. Séby, Cr(VI) speciation in foods by HPLC-ICP-MS: investigation of Cr(VI)/food interactions by size exclusion and Cr(VI) determination and stability by ion-exchange on-line separations, Anal. Bioanal. Chem., 2015, 407, 3831–3839 CrossRef CAS PubMed.
- S. Saputro, K. Yoshimura, S. Matsuoka, K. Takehara, J. Aizawa and Y. Tennichi, Speciation of dissolved chromium and the mechanisms controlling its concentration in natural water, Chem. Geol., 2014, 364, 33–41 CrossRef CAS.
- O. Senofonte and F. Petrucci, Determination of Cr(VI) in cosmetic products using ion chromatography with dynamic reaction cell-inductively coupled plasma-mass spectrometry (DRC-ICP-MS), Anal. Methods, 2015, 7, 5269–5274 RSC.
- T. H. Ding, H. H. Lin and C. W. Whang, Determination of chromium(III) in water by solid-phase microextraction with a polyimide-coated fiber and gas chromatography-flame photometric detection, J. Chromatogr. A, 2005, 1062, 49–55 CrossRef CAS PubMed.
- N. Jie, Q. Zhang, J. Yang and X. Huang, Determination of chromium in waste-water and cast iron samples by fluorescence quenching of rhodamine 6G, Talanta, 1998, 46, 215–219 CrossRef CAS PubMed.
- G. A. O. Jia-you, Determination of chromium(VI) with rhodamine 6G by fluorescence quenching method, Metall. Anal., 2005, 1, 009 Search PubMed.
- T. Madrakian, A. Afkhami and M. Mohammadnejad, Application of organized media for rapid spectrofluorimetric determination of trace amounts of Cr(VI) in the presence of Cr(III), Bull. Korean Chem. Soc., 2009, 30, 1252–1256 CrossRef CAS.
- H. Y. Kuang, X. Y. Yuan, L. Feng, H. W. Huang, C. R. Tang, K. Q. Deng and Y. L. Zeng, Simple and sensitive detection for trace chromium(VI) using albumin as fluorescence probe, J. Chil. Chem. Soc., 2015, 60, 2840–2842 CrossRef CAS.
- M. S. El-Shahawi, H. M. Al-Saidi, A. S. Bashammakh, A. A. Al-Sibaai and M. A. Abdelfadeel, Spectrofluorometric determination and chemical speciation of trace concentrations of chromium(III & VI) species in water using the ion pairing reagent tetraphenyl-phosphonium bromide, Talanta, 2011, 84, 175–179 CrossRef CAS PubMed.
- V. Mahesvari and N. Balasabramanian, Spectrophotometric determination of chromium based on ion-pair formation, Chem. Anal., 1996, 41, 569–576 Search PubMed.
- M. Alexovič, I. S. Balogh, J. Škrlíková and V. A. Andruch, A dispersive liquid–liquid microextraction procedure for UV-vis spectrophotometric determination of chromium(VI) in water samples, Anal. Methods, 2012, 4, 1410–1414 RSC.
- M. D. M. Abadi, M. Chamsaz, M. H. Arbab-Zavar and F. Shemirani, Supramolecular dispersive liquid–liquid microextraction based solidification of floating organic drops for speciation and spectrophotometric determination of chromium in real samples, Anal. Methods, 2013, 5, 2971–2977 RSC.
- M. S. El-Shahawi, S. S. M. Hassan, A. M. Othman, M. A. Zyada and M. A. El-Sonbati, Chemical speciation of chromium(III,VI) employing extractive spectrophotometry and tetraphenylarsonium chloride or tetraphenylphosphonium bromide as ion-pair reagent, Anal. Chim. Acta, 2005, 534, 319–326 CrossRef CAS.
- M. Telepčáková, V. Andruch and I. S. Balogh, Indirect extraction-spectrophotometric determination of chromium, Chem. Pap., 2005, 59, 109–112 Search PubMed.
- Z. Kong, J. Wei, H. Guo, H. Wang, Y. Li and N. Liu, The determination of trace amount of Cr(VI) and As(V) in aqueous solution by UV–vis spectrophotometer assisted with preconcentration on a quaternary ammonium ion exchange fiber-in-tube, Desalin. Water Treat., 2015, 53, 382–389 CrossRef CAS.
- D. A. Goncalves, J. Gu, M. C. dos Santos, B. T. Jones and G. L. Donati, Direct determination of chromium in empty medicine capsules by tungsten coil atomic emission spectrometry, J. Anal. At. Spectrom., 2015, 30, 1395–1399 RSC.
- Y. Yang, C. Y. Gao, J. Liu and D. Dong, Recent developments in rhodamine salicylidene hydrazone chemosensors, Anal. Methods, 2016, 8, 863–2871 Search PubMed.
- R. K. Tandon, P. T. Crisp, J. Ellis and R. S. Baker, Effect of pH on chromium(VI) species in solution, Talanta, 1984, 31, 227–228 CrossRef CAS PubMed.
- I. S. Balogh, I. M. Maga, A. Hargitai-Toth and V. Andruch, Spectrophotometric study of the complexation and extraction of chromium(VI) with cyanine dyes, Talanta, 2000, 53, 543–549 CrossRef CAS PubMed.
- C. E. Crouthamel and C. E. Johnson, Thiocyanate spectrophotometric determination of molybdenumand tungsten, Anal. Chem., 1954, 26(8), 1284–1291 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, ( 2013) Search PubMed.
- S. A. El-Daly, S. A. El-Azim, F. M. Elmekawey, B. Y. Elbaradei, S. A. Shama and A. M. Asiri, Photophysical parameters, excitation energy transfer, and photoreactivity of 1,4-bis(5-phenyl-2-oxazolyl)benzene (POPOP) laser dye, Int. J. Photoenergy, 2012, 2012, 1–10 CrossRef.
- V. K. Bhardwaj, A. P. S. Pannu, N. Singh, M. S. Hundal and G. Hundal, Synthesis of new tripodal receptors—a ‘PET’ based ‘off–on’ recognition of Ag+, Tetrahedron, 2008, 64, 5384–5391 CrossRef CAS.
- J. C. Miller and J. N. Miller, Statistics for Analytical Chemistry, Ellis Horwood Limited, New York, 4th edn, ( 1994) Search PubMed.
Footnote |
| † On sabbatical leave from Department of Chemistry, Faculty of Science, Damietta University, Damietta, Egypt. |
| This journal is © The Royal Society of Chemistry 2016 |