
Frustratingly synergic effect of cobalt–nickel heterometallic precatalysts on ethylene reactivity: the cobalt and its heteronickel complexes bearing 2-methyl-2,4-bis(6-aryliminopyridin-2-yl)-1H-1,5-benzodiazepines†
Abstract
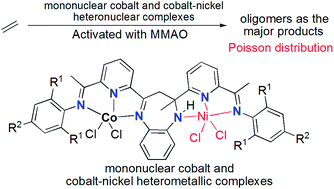
A series of 2-methyl-2,4-bis(6-aryliminopyridin-2-yl)-1H-1,5-benzodiazepines were reacted with cobalt chloride to form their corresponding cobalt complexes, which were further reacted with nickel chloride to form their binuclear heteronickel complexes. One of the representative cobalt–nickel complexes was unambiguously confirmed using single-crystal X-ray diffraction. Generally all mononuclear cobalt complexes and binuclear cobalt–nickel complexes showed good activities towards ethylene reactivity (oligomerization and polymerization), however, the mononuclear cobalt complexes showed much higher activities than the binuclear cobalt–nickel analogues, and this is described as the frustratingly synergic effect of heterometallic complexes in ethylene oligomerization. In addition, the obtained oligomers sometimes followed a Poisson distribution.
 Please wait while we load your content...
Please wait while we load your content...

