DOI:
10.1039/C6RA13045C
(Paper)
RSC Adv., 2016,
6, 73581-73588
Enhancing oxygen reduction reaction durability via coating graphene layers on iron-nitrogen supported carbon nanotubes†
Received
19th May 2016
, Accepted 29th July 2016
First published on 29th July 2016
Abstract
Development of efficient, low-cost and good stability electrocatalysts as alternatives to platinum for the oxygen reduction reaction (ORR) is of significance for fuel cells. Here we report a novel type of Fe, N supported carbon nanotube encapsulated with nitrogen doped graphene as an ORR electrocatalyst synthesized by a hydrothermal method, for which nitrogen-enriched melamine, ferric chloride and disodium edentate were used as the nitrogen, iron and graphene precursors, respectively. Disodium edentate plays an important role in the performance towards the ORR because it is not only a graphene precursor, but also the key of forming FeNx active sites. The prepared Fe-N-CNT@GN catalyst exhibits high ORR activity in alkaline media with an onset potential of −0.13 V, a limiting current density of 6.2 mA cm−2, and higher selectivity (number of electron transfer n ∼ 3.8) which might be due to the FeNx active sites. Moreover, the half-wave potential exhibits almost no changes after 8000 continuous cycles scanning from −0.3 V to 0.1 V. The tolerance to the methanol crossover effect in alkaline media is superior. The Fe-N-CNT@GN catalyst obtained by this method solves the problem of agglomeration, migration, dissolution and leaching of catalysts in alkaline media. This type of catalyst has great potential for use as a high-performance nonprecious metal cathode catalyst in PEMFCs.
1. Introduction
The development and utilization of renewable energy has become a global concerned focus because of the depletion of fossil resources and environmental degradation.1 Proton exchange membrane fuel cells (PEMFCs) have emerged as leading candidates in the alternative energy industry.2,3 However, the sluggish kinetics of the oxygen reduction reaction (ORR) results in serious cathode polarization and energy loss, which restrict their large-scale application. Pt-based catalysts have been proven to be the most efficient catalysts for the ORR. However, the prohibitive cost and scarcity of Pt significantly hinder their commercialization.4–6 Additionally, the durability of Pt is poor which is often caused by different degradation mechanisms during long-term electrochemical operation, such as nanoparticle migration, Ostwald ripening, coalescence and leaching.7,8
In quest of practical catalysts with lower price and comparative or even better catalytic performance than Pt-based catalysts, non-precious metal catalysts (NPMCs),9 such as transition metal macrocyclic compounds10 and transition metal oxide,11 have been devoted great efforts. Among the key breakthroughs in this area were those by Jasinski et al., who were the first to report the oxygen reduction capabilities of cobalt phthalocyanine in basic media.12 Followed by pyrolysis of transition metal macrocyclic compounds (e.g. cyanocobalamin,13–15 porphyrin,16,17and phthalocyanine18) have been demonstrated to be an effective method to achieve highly active and durable catalysts. However, these transition metal macrocyclic compounds are generally expensive, even comparable to Pt/C catalyst, which significantly hinders their scalable production. Transition metal oxide catalysts were reported exhibiting high activity for ORR,19–22 however, also frequently suffer from dissolution and agglomerations during the operation of fuel cells, which may result in severe catalyst degradation.23 Deng et al.,24 reported a bottom-up method for the preparation of ultrathin graphene spheres with only one to three graphene layers that encapsulate CoNi nanoalloy electrocatalysts for hydrogen evolution reaction. This strategy represents a new concept for maintaining the stability of non-precious metals in electrolyte solution. But the large size metal nanoparticles resulting from agglomeration decreases the electrochemical specific surface area, which goes against the improvement of the utilization for the catalysts.
In this work, a novel type of Fe, N supported carbon nanotubes (CNTs) encapsulated with graphene layers was developed through hydrothermal method, using nitrogen-enriched melamine, ferric chloride and disodium edentate as nitrogen, iron and graphene precursors, respectively. CNTs exhibit outstanding advantages, such as large surface area, good electrical conductivity and more adsorption active sites, which is favorable to synthesis catalysts with high metal dispersion. Different with the reported method that directly coating the metal with graphene layers,25 iron and nitrogen elements were firstly supported on the carboxyl group functioned CNTs. Then graphene layers were coated with hydrothermal and subsequent calcination methods which was denoted as Fe-N-CNT@GN. The obtained Fe-N-CNT@GN exhibits high selectivity for the ORR, long-term operation durability in alkaline solution and outstanding immunity to methanol crossover. This simple and economic method solves the problem of metal nanoparticles agglomeration and offers an alternative route to improve the durability of precious-metal-free catalysts.
2. Experimental details
2.1 Materials
Carbon nanotubes (XFM09) are obtained from XF NANO. FeCl3·6H2O (≥99.0%) is purchased from DAMAO. Melamine (99.5%) is purchased from Guangfu Chemical Reagents. Disodium edentate (99.0%), methanol (99.5%), and ethanol (99.7%) are purchased from Kermel. Nafion (5%) is purchased from Sigma-Aldrich. These chemicals are used as received without further purification. Deionized water through Smart system (Smart-Q15) is used in all experiments.
2.2 Catalysts preparation
The Fe-N-CNT@GN catalyst was synthesized by hydrothermal synthesis method, as illustrated in Fig. 1. In a typical synthesis, 0.100 g of carboxyl carbon nanotubes, 0.0386 g of FeCl3·6H2O (mass ratio of Fe element/CNTs is 8 wt%) and 0.125 g of melamine were dissolved/dispersed uniformly in 30 mL mixed solution of ethanol and water (volume ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) by ultrasonication for 0.5 h, and followed by stirred at 50 °C for 2 h. Then, the obtained mixture, a certain amount of disodium edentate (C10H16N2Na2O8) and 10 mL of methanol were together enclosed into a 100 mL stainless steel autoclave and followed by processing at 150 °C for 6 h. After cooling to room temperature naturally, the generated precipitates were filtered and washed with deionized water, and finally dried under vacuum at 80 °C overnight. The obtained compound was denoted as Fe-N-CNT-EDTA. After that, Fe-N-CNT-EDTA compound was heat-treated at a temperature of 900 °C for 1 h in N2 flow to obtain Fe-N-CNT@GN catalyst. For comparison, the preparation of Fe-CNT, N-CNT and Fe-N-CNT, a mixture solution of CNTs (0.100 g) and FeCl3·6H2O (0.0386 g), a mixture solution of CNTs (0.100 g) and melamine (0.125 g), a mixture solution of CNTs (0.100 g), FeCl3·6H2O (0.0386 g) and melamine (0.125 g) was respectively used, all other conditions were the same as those for the Fe-N-CNT@GN.
:1) by ultrasonication for 0.5 h, and followed by stirred at 50 °C for 2 h. Then, the obtained mixture, a certain amount of disodium edentate (C10H16N2Na2O8) and 10 mL of methanol were together enclosed into a 100 mL stainless steel autoclave and followed by processing at 150 °C for 6 h. After cooling to room temperature naturally, the generated precipitates were filtered and washed with deionized water, and finally dried under vacuum at 80 °C overnight. The obtained compound was denoted as Fe-N-CNT-EDTA. After that, Fe-N-CNT-EDTA compound was heat-treated at a temperature of 900 °C for 1 h in N2 flow to obtain Fe-N-CNT@GN catalyst. For comparison, the preparation of Fe-CNT, N-CNT and Fe-N-CNT, a mixture solution of CNTs (0.100 g) and FeCl3·6H2O (0.0386 g), a mixture solution of CNTs (0.100 g) and melamine (0.125 g), a mixture solution of CNTs (0.100 g), FeCl3·6H2O (0.0386 g) and melamine (0.125 g) was respectively used, all other conditions were the same as those for the Fe-N-CNT@GN.
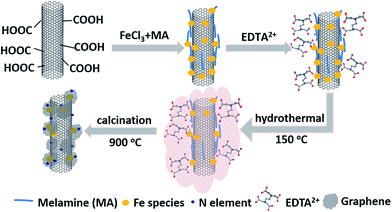 |
| | Fig. 1 Schematic illustration of the synthesis of Fe-N-CNT@GN catalyst. | |
2.3 Characterization
Scanning electron microscopy (SEM) was conducted on Hitachi S-4800 operated at 30 kV. Transmission Electron Microscopy (TEM) images were examined on a Tecnai G2 F20 microscope at 200 kV. X-ray diffraction (XRD) patterns were recorded on a Shimazduo XRD-7000S diffractometer with Cu Kα radiation (λ = 1.54 Å), and the scanning rate was 5° min−1, and the tube voltage and current were maintained at 40 kV and 200 mA. X-ray photoelectron spectroscopy (XPS) datas were obtained on a Kratos AMICUS instruments operated at 25 W and 1486.6 eV with monochromated Al Kα radiation, and binding energies were corrected with reference to the C 1s peak at 284.8 eV.
2.4 Electrochemical measurements
All electrochemical performances for the Fe-N-CNT@GN were performed on an electrochemical analyzer (CHI660E) with a three-electrode system. A Pt wire works as counter electrode, a KCl-saturated Ag/AgCl as reference electrode and a glassy carbon electrode (GCE) supported with a catalyst film as working electrode (disk area of 0.0707 cm2). The working electrode was prepared as follows: the GCE was first polished with 50 nm alumina powders and then washed in deionized water to get a mirror-like surface. 20 μL of the as-prepared catalysts ink was pipetted onto the GCE and dried at room temperature to obtain the working electrode. Accordingly, the catalyst loading is 0.566 mg cm−2. To get the as-prepared catalyst ink, 4 mg of the corresponding catalyst with 2 mL of ethanol was first ultrasonically mixed for 15 minutes. Then 30 μL of 5 wt% Nafion was added to and the ink was ultrasonically stirred for another 15 minutes. All potentials in this work are referred to KCl-saturated Ag/AgCl electrode. To test the ORR activity, cyclic voltammetry (CV) characterization of Fe-N-CNT@GN was carried out in O2-saturated 0.1 M KOH electrolyte in the potential range from −0.8 to 0.2 V at a scan rate of 10 mV s−1 and 1600 rpm electrode rotation speed. To correct the background current, the electrochemical characterization of the catalysts in N2-saturated 0.1 M KOH was also recorded (the ORR curves were corrected by subtracting the voltammogram recorded in N2-satarated electrolyte). The durability tests of the Fe-N-CNT@GN were performed with two methods: (a) the chronoamperometric current–time responses of Fe-N-CNT@GN in O2-saturated 0.1 M KOH under the potential of −0.4 V for 2400 s at the electrode rotation speed of 1600 rpm, (b) the accelerated aging tests (AATs) of the Fe-N-CNT@GN which were performed in O2-saturated 0.1 M KOH at the potential range from −0.3 V to 0.1 V with a scan rate of 100 mV s−1 at 1600 rpm. The anti-methanol tests of the Fe-N-CNT@GN were performed in O2-saturated 0.1 M KOH with 3 M methanol solution. For comparison, the commercial Pt/C (20 wt%, Alfa Aesar) was also tested in the same conditions. In addition, rotating ring disk electrode (RRDE) measurements of Fe-N-CNT@GN catalyst was conducted in a three-electrode cell setup with a computer controlled bipotentiostat (Pine Company). 40 μL of the catalyst ink was pipetted on the surface of the GCE (Ø 5.7 mm, with a Pt ring for RRDE) to form a catalyst loading of 0.313 mg cm−2. In this work, the current density was obtained from this equation: J = (IO2 − IN2) × 1000/SGCE, where J is current density, IO2 and IN2 are current in O2- and N2-saturated 0.1 M KOH electrolyte, respectively, SGCE is the area of the GCE for RDE or RRDE.
3. Results and discussion
3.1 Structural characterization of Fe-N-CNT@GN
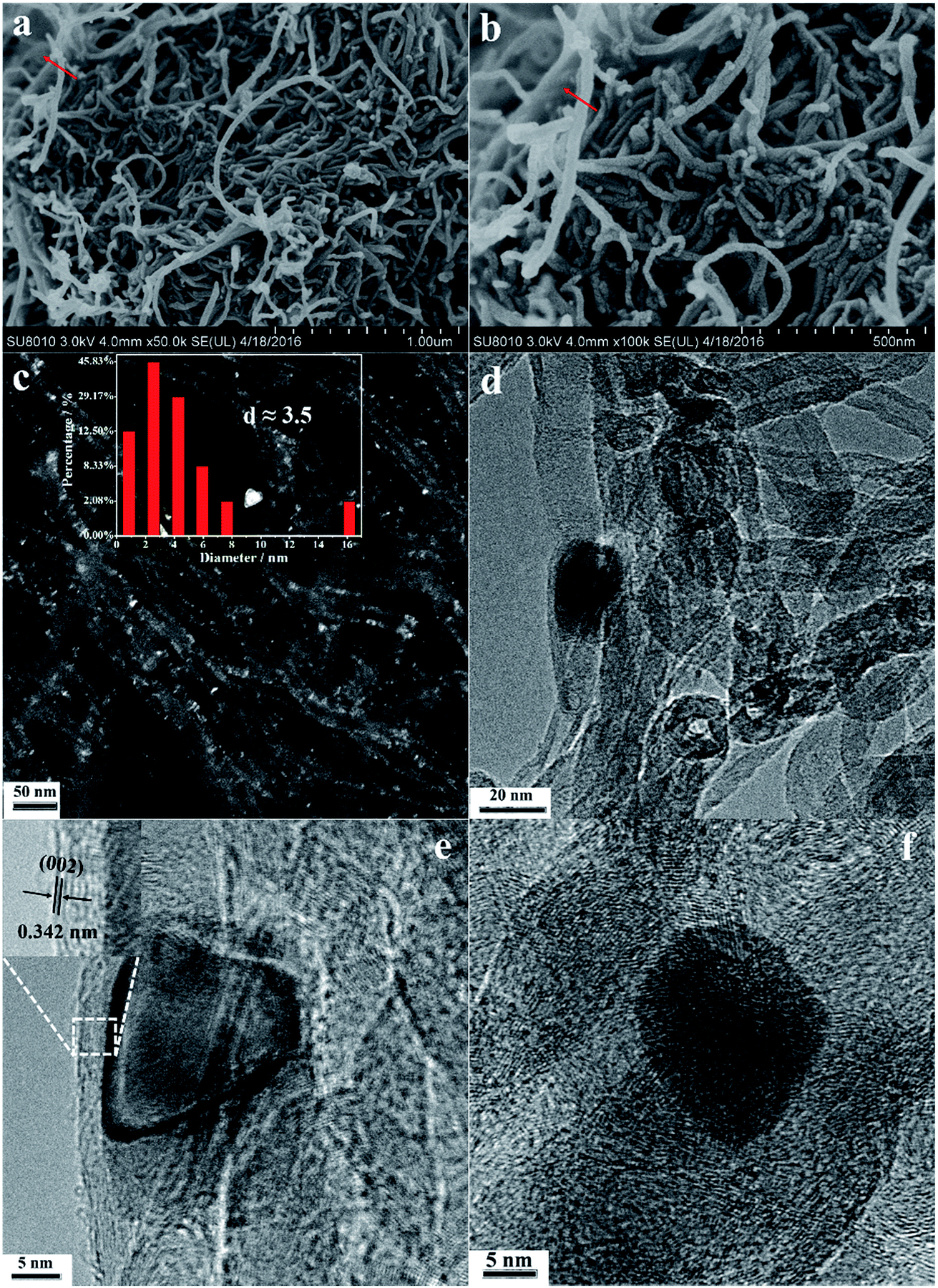
SEM images (Fig. 2a and b, the red arrows designate the layered graphene sheets in each image) showed that the Fe-N-CNT@GN catalyst consists of the winding CNTs together and the forming lamellar graphene sheets. Moreover, few metal nanoparticles have been found on the surface of Fe-N-CNT@GN from the SEM images because metal nanoparticles may be coated by the graphene sheets formed on CNTs, which was further studied by the dark field transmission electron microscopy (DFTEM) as illustrated in Fig. 2c. It can be seen that a large quantity of Fe nanoparticles dispersed highly on the CNTs with an average particle diameter of about 3.5 nm. High-resolution TEM (HRTEM) showed the typical structure that Fe nanoparticles were coated by several graphite-like layers (Fig. 2d), which could easily be identified with an interlayer spacing of 0.342 nm (the top-left inset of Fig. 2e), corresponding to the (002) plane of graphitic carbon. Furthermore, as depicted in Fig. 2f, the lattice fringes of Fe nanoparticles, CNT and graphene layers are overlapping with each other, which makes the plane of Fe could not be identified. Therefore, the crystal structure of Fe-N-CNT@GN was studied by XRD patterns. For comparison, the CNT, Fe-CNT and Fe-N-CNT were also tested as shown in Fig. 3. It is clear that all catalysts exhibit the (002) carbon plane diffraction peak at 26.4°, which is mainly from the CNTs. It should be noted that the peak intensities of the Fe-N-CNT@GN are clearly higher than those of the Fe-CNT and Fe-N-CNT. This may reflect the stronger graphitization degree of the Fe-N-CNT@GN catalyst with the assistance of graphene layers. The peaks at 54.4° and 78.6° of the Fe-N-CNT@GN can be attributed to the (230), (133) planes of Fe3C (PCPDF # 35-0772), and the weak peaks at around 43.4° and 63.7° indicate the formation of Fe3N crystalline (PCPDF # 49-1662). Different with Fe-N-CNT@GN, the XRD patterns of the Fe-CNT and Fe-N-CNT catalysts show the C0.09Fe1.91 diffraction peaks at 43.9°, 44.9°, 65.3° and 82.1° (PCPDF # 44-1292) respectively, indicating no Fe3N and Fe3C was formed. In terms of the preparation of Fe-N-CNT@GN, EDTA2+ is a widely used chelating agent in solution chemistry,26 but here it is used as a graphene source. Fe-N-CNT-EDTA was synthesized by hydrothermal method. During the calcination process, Fe-N-CNT-EDTA will be decomposed and generate CNx species which were more likely to transfer to graphene layers around iron nanoparticles. Fe–Nx–C bonds might be formed by hydrothermal and calcination process due to the strong coordination capability of EDTA2+. FeNx species is beneficial to the performance for ORR.27–29 The sizes of Fe nanoparticles for Fe-N-CNT@GN catalysts obtained from the XRD data according to the Scherrer formula is about 3.62 nm, which is close to the TEM result. But the Fe nanoparticles sizes of the Fe-CNT and Fe-N-CNT catalysts are about 4.55 nm, 4.43 nm respectively. The above results suggest that Fe element in Fe-N-CNT@GN catalyst has been supported on CNTs and combined to nitrogen element forming the Fe3N structure, which may provide active sites for ORR.
 |
| | Fig. 2 SEM images of Fe-N-CNT@GN (a and b), DFTEM image of Fe-N-CNT@GN (c), HRTEM images of Fe-N-CNT@GN (d and f). | |
 |
| | Fig. 3 XRD patterns of Fe-N-CNT@GN, Fe-N-CNT, Fe-CNT and CNT. | |
XPS was used to analyze the elemental chemical states and contents of the catalysts which would directly affect the electrochemical performance. In order to analyze the effect of iron, nitrogen elements on the activity of the Fe-N-CNT@GN, CNT, Fe-CNT, N-CNT and Fe-N-CNT were also investigated for comparison, as shown in Fig. S1†. The contents of C, N, O and Fe of all the above catalysts derived from Fig. S1 and S3a† are summarized in Table S1.† Comparing to the N content of N-CNT (0.7 at%) and Fe-N-CNT (0.6 at%), it is clear that the total nitrogen content of the Fe-N-CNT@GN is highest (total N content of Fe-N-CNT@GN is 0.9 at%) because the graphene precursor EDTA2+ salt also offer high content nitrogen source. For another aspect, the Fe content of Fe-N-CNT@GN (0.2 at%) is much lower than that of the Fe-N-CNT (0.4 at%) and Fe-CNT (0.4 at%). Considering the amount of all reactants for synthesizing Fe-CNT, Fe-N-CNT and Fe-N-CNT@GN is identical, it may deduce that the Fe nanoparticles are coated with graphene layers synthesized from EDTA2+ salt. In order to get more detailed information, the overlapped high-resolution C 1s peak of Fe-N-CNT@GN was decomposed into two components (Fig. S3b†), which were arising from sp2 hybridized C (284.7 eV), C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (285.4 eV),30,31 which represented that nitrogen elements have been incorporated into carbon, and the high content of sp2 C (about 45.9 at% in total C species) would lead to high electrical conductivity and hence high ORR activity. As depicted in Fig. S3c,† a trivial N 1s signal was observed for Fe-N-CNT@GN (mainly from pyridinic N and pyrrolic N),33 signifying a negligible share of nitrogen atoms on the Fe-N-CNT@GN catalyst surface, although EDTA2+ salt was added which might partial decompose during the calcination step, let alone the N-CNT and Fe-N-CNT catalysts. In addition, it also can be easily observed that a very poor signal/noise ratio revealed a negligible surface iron content on the Fe-N-CNT@GN surface from Fig. S3d.† Considering the Fe content is 8 wt%, it can be deduced that the low Fe contents of Fe-N-CNT@GN detected by XPS is due to the Fe is embedded in graphene layers which screen the XPS signal.32,33 These may be the reason that our catalyst Fe-N-CNT@GN presents a superior ORR durability as described below. As to the Fe-CNT and Fe-N-CNT catalysts, the XPS spectra of Fe can be further deconvoluted into three different species: FeII 2p3/2 about 710 eV, FeIII 2p3/2 about 713 eV and FeIII 2p1/2 about 725 eV which are consistent with previously reported spectra for Fe-containing species doped into carbon materials.29,34,35
N (285.4 eV),30,31 which represented that nitrogen elements have been incorporated into carbon, and the high content of sp2 C (about 45.9 at% in total C species) would lead to high electrical conductivity and hence high ORR activity. As depicted in Fig. S3c,† a trivial N 1s signal was observed for Fe-N-CNT@GN (mainly from pyridinic N and pyrrolic N),33 signifying a negligible share of nitrogen atoms on the Fe-N-CNT@GN catalyst surface, although EDTA2+ salt was added which might partial decompose during the calcination step, let alone the N-CNT and Fe-N-CNT catalysts. In addition, it also can be easily observed that a very poor signal/noise ratio revealed a negligible surface iron content on the Fe-N-CNT@GN surface from Fig. S3d.† Considering the Fe content is 8 wt%, it can be deduced that the low Fe contents of Fe-N-CNT@GN detected by XPS is due to the Fe is embedded in graphene layers which screen the XPS signal.32,33 These may be the reason that our catalyst Fe-N-CNT@GN presents a superior ORR durability as described below. As to the Fe-CNT and Fe-N-CNT catalysts, the XPS spectra of Fe can be further deconvoluted into three different species: FeII 2p3/2 about 710 eV, FeIII 2p3/2 about 713 eV and FeIII 2p1/2 about 725 eV which are consistent with previously reported spectra for Fe-containing species doped into carbon materials.29,34,35
3.2 Electrochemical activity of Fe-N-CNT@GN catalyst towards ORR
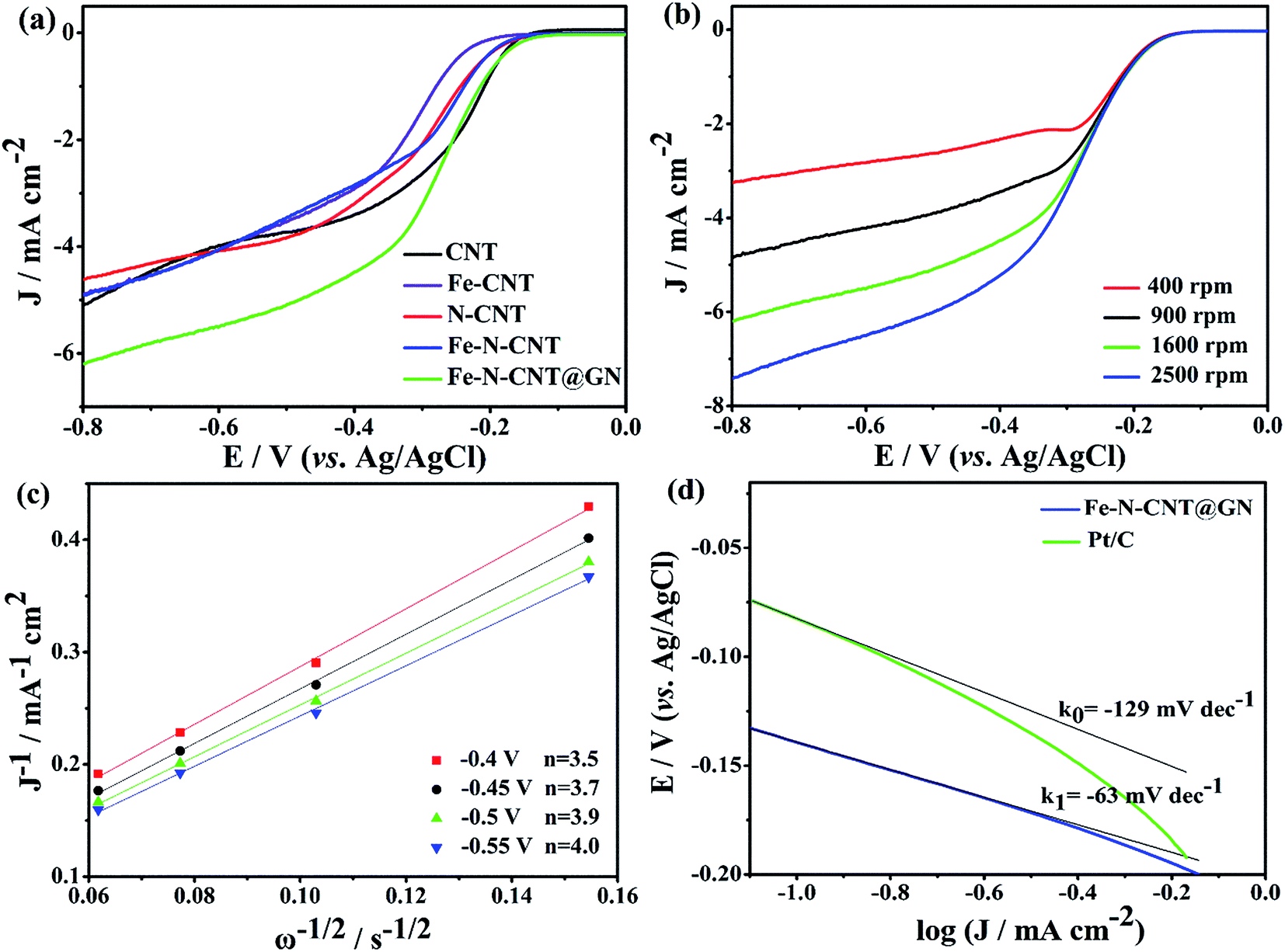
3.2.1 The performance of ORR. Cyclic voltammetry (CV) was employed to compare the catalysts electrochemical characterization in N2- or O2-saturated 0.1 M KOH solution. As shown in Fig. S4,† the CV curves of Fe-N-CNT@GN are essentially featureless in the N2-saturated solution, whereas the catalyst exhibits significant cathodic ORR peaks in the O2-saturated electrolyte, suggesting a pronounced electrocatalytic activity of the Fe-N-CNT@GN. Furthermore, the ORR catalytic performance of Fe-N-CNT@GN was compared with CNT, Fe-CNT, N-CNT and Fe-N-CNT as shown in Fig. 4a, where the current density at −0.4 V of these catalysts increase significantly in the following order: Fe-CNT < Fe-N-CNT < N-CNT < CNT < Fe-N-CNT@GN. And the Fe-N-CNT@GN catalyst displays the most positive ORR onset potential of −0.13 V and the highest limiting current density of 6.2 mA cm−2. The ORR activity of Fe-N-CNT@GN was also compared with that of the commercial Pt/C catalyst (20 wt%, Fig. S5†). The half-wave potential of Fe-N-CNT@GN (−0.25 V) is only slightly more negative than 20 wt% Pt/C (−0.16 V), while the limiting current density is even higher than that of the Pt/C below −0.56 V. The possible reason is that the surface of Fe-N-CNT@GN is beneficial to the form of ORR three phase interfaces, which accelerates diffusion of reactants and products.
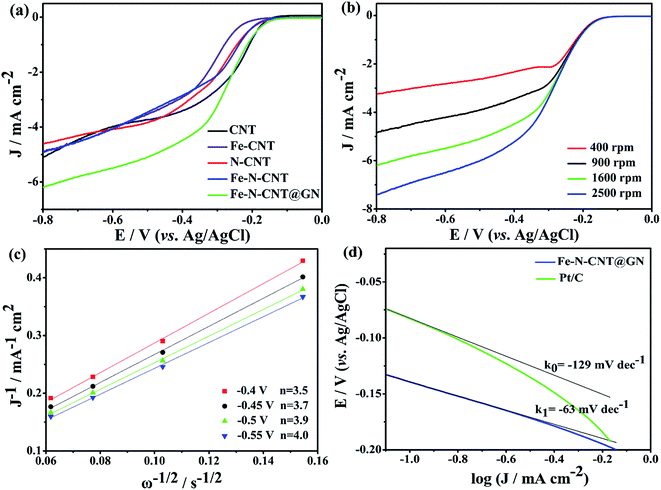 |
| | Fig. 4 (a) CV curves of CNT, Fe-CNT, N-CNT, Fe-N-CNT, Fe-N-CNT@GN samples in O2-saturated 0.1 M KOH solution, scan rate: 10 mV s−1, rotation rate: 1600 rpm, J = (IO2 − IN2) × 1000/0.0707. (b) CV curves of Fe-N-CNT@GN in O2-saturated 0.1 M KOH solution at different rotation rates. (c) Koutecky–Levich plots of Fe-N-CNT@GN derived from CV curves in (b) at different electrode potentials. (d) The Tafel plots of the Fe-N-CNT@GN and Pt/C catalysts derived from their CV data in Fig. S5.† | |
To further investigate the pathway and kinetics process of the ORR, polarization curves of the Fe-N-CNT@GN catalyst was recorded using the RDE technique at different rotation rates (400, 900, 1600 and 2500 rpm) in O2-saturated electrolyte with a scan rate of 10 mV s−1 (Fig. 4b). According to the ORR polarization curves at various rotation rates, the corresponding Koutecky–Levich plots (Fig. 4c) were obtained and exhibited good linearity with an almost constant slope at electrode potentials ranging from −0.8 V to −0.4 V, indicating that the ORR process follows first-order kinetics with respect to the concentration of dissolved O2.36,37 The electron transfer number (n) during the ORR can be calculated according to the Koutecky–Levich eqn (1)–(3):19,38
| | |
JL = 0.62nFD2/3ν−1/6ω1/2c0
| (2) |
where
J is the current density,
JK and
JL are the kinetic- and diffusion-limiting current densities, respectively,
ω is the angular velocity of the disk (
ω = 2π
N, where
N is the RDE rotation speed),
n is the number of electrons transferred in the ORR,
F is the Faraday constant (96
485 C mol
−1),
c0 is the bulk concentration of O
2 (1.2 × 10
−6 mol cm
−3 in 0.1 M KOH solution),
ν is the kinematic viscosity of the electrolyte (1 × 10
−2 cm
2 s
−1 in 0.1 M KOH solution),
D is the diffusion coefficient of O
2 in 0.1 M KOH solution (1.9 × 10
−5 cm
2 s
−1) and
k is the electron transfer rate constant.
37 As shown in
Fig. 4c and S6i,
† the calculated average
n value for the Fe-N-CNT@GN (3.8) is close to 4, which is higher than that for the CNT (3.0), Fe-CNT (3.2), N-CNT (3.5) and Fe-N-CNT (3.1) catalysts between −0.8 V and −0.4 V. Such results indicate that the ORR on the Fe-N-CNT@GN electrocatalyst performs good selectivity toward the four-electron pathway of ORR with H
2O as the main product during the cathodic reaction. The excellent ORR catalytic activity of Fe-N-CNT@GN was also gleaned from a smaller Tafel slope of 63 mV dec
−1 at the low over-potentials (
Fig. 4d). The high selectivity for the four-electron transfer pathway of Fe-N-CNT@GN can be attributed to the synergistic activity sites created by doped N and Fe species.
39
To further verify the ORR catalytic efficiency of the Fe-N-CNT@GN catalyst, we also conducted the RRDE measurements at 10 mV s−1 with a rotation speed of 1600 rpm. The n values were calculated from eqn (4) and the HO2− formation yields were calculated from eqn (5) based on the RRDE data:5
| | |
% HO2− = 200IR/(IDN + IR)
| (5) |
where
ID is the disk current,
IR is the ring current, and
N is the current collector efficiency of the Pt ring (0.38 for this work). The
IR and
ID values for the Fe-N-CNT@GN catalyst was shown in
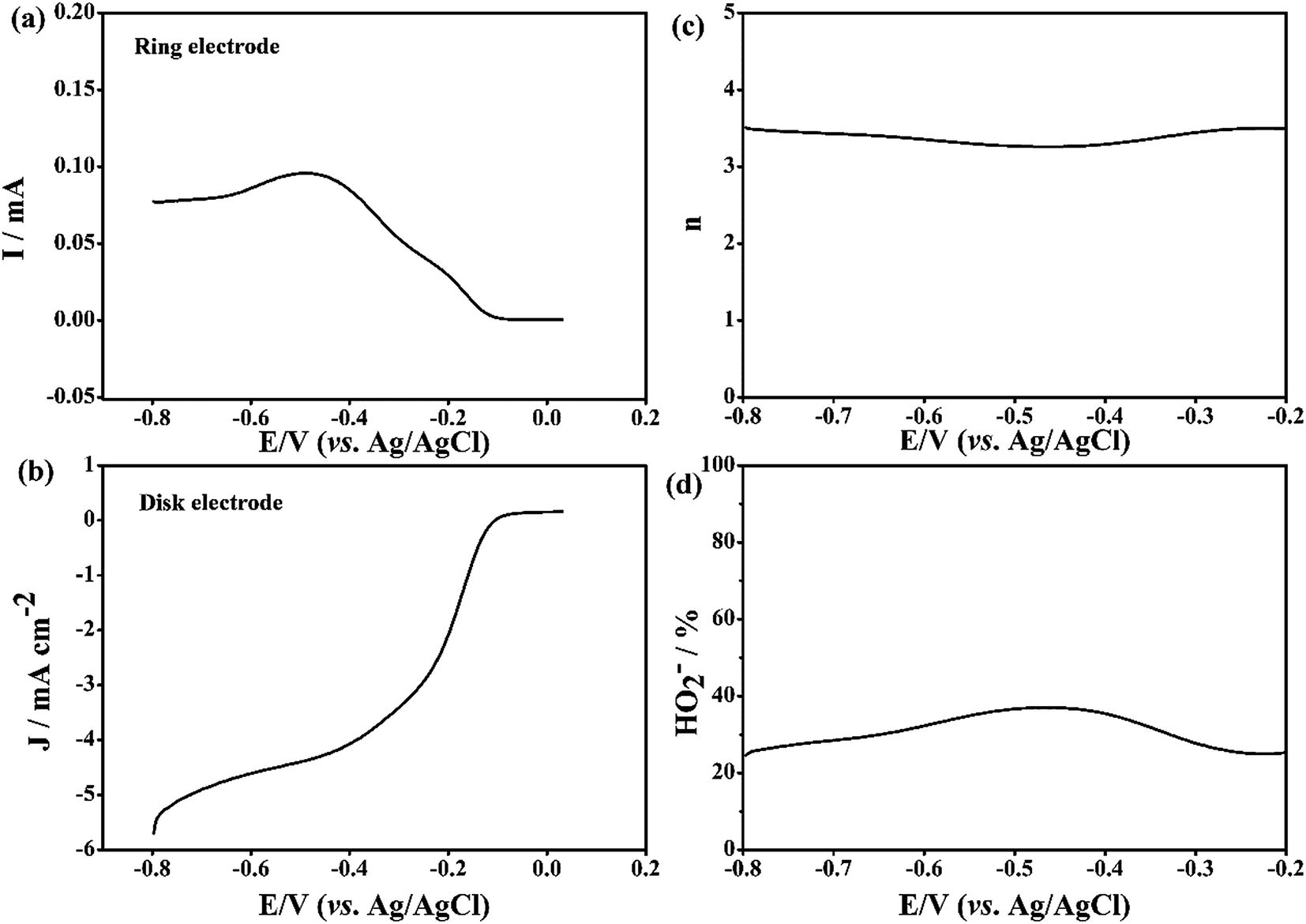
Fig. 5a and b, respectively. It is clear that the calculated
n values are of about 3.7 for the Fe-N-CNT@GN catalysts (
Fig. 5c). This is nearly consistent with the results obtained from the K–L plots based on the RDE measurements. At the same time, the measured HO
2− yields for the Fe-N-CNT@GN are below 36% over the potential range of −0.8 to −0.2 V (
Fig. 5d). Therefore, the RRDE measurements also demonstrate that oxygen is reduced through the dominant four electron process. All electrochemical experiments indicate that the Fe-N-CNT@GN catalyst is a good alternative to precious metals as a cathode catalyst.
 |
| | Fig. 5 (a) Ring current, (b) disk current density, (c) the electron transfer numbers n and (d) HO2− yields of the Fe-N-CNT@GN in O2-saturated 0.1 M KOH by the RRDE technique. Scan rate: 10 mV s−1, rotation rate: 1600 rpm, electrode area: 0.255 cm2, Jdisk = (IO2 − IN2) × 1000/0.255. | |
3.2.2 Durability and tolerance to the methanol crossover effect. The electrocatalytic durability and tolerance to methanol crossover effect of the ORR catalyst at the cathode are major concerns in practical applications of fuel cells.40 In Fig. 6, the chronoamperometric current–time responses of Fe-N-CNT@GN at −0.4 V in O2-saturated 0.1 M KOH solution at 1600 rpm exhibits a very good durability with a high relative current density of 95% of its initial current density after 2400 s, whereas the Pt/C exhibits a more rapid attenuation with a current loss of 17%. Furthermore, the stabilities of the Fe-N-CNT@GN and Pt/C were also evaluated by an accelerated aging test as shown in the Fig. S7.†41,42 After 8000 continuous cycles scanning from −0.3 V to 0.1 V in O2-saturated 0.1 M KOH at 1600 rpm, the half-wave potential of the Fe-N-CNT@GN exhibits almost no negative shift under O2, while the negative shift of ΔE1/2 of Pt/C is about 20 mV, validating the high durability of the Fe-N-CNT@GN catalyst. Such outstanding stability of the Fe-N-CNT@GN catalyst might be ascribed to not only the support role of carbon nanotubes but also the formation of a nitrogen doped graphene layers that could retard their dissolution and leach in the alkaline media during long-term operation. Combining to the XPS and ORR results, it can be deduced that FeNx active sites which encapsulated with the N-doped graphene layers might enhance the durability of the Fe-N-CNT@GN catalyst.
 |
| | Fig. 6 Chronoamperometric current–time responses of the Fe-N-CNT@GN and Pt/C catalysts on the RDE kept at −0.4 V (vs. Ag/AgCl) in O2-saturated 0.1 M KOH, rotation rate: 1600 rpm. | |
Fe-N-CNT@GN and commercial Pt/C catalysts were further compared by introducing methanol into the electrolyte to examine the catalysts selectivity and anti-crossover effects. Fig. 7 shows CV curves of Fe-N-CNT@GN and commercial Pt/C catalysts in O2-saturated 0.1 M KOH solution and O2-saturated 0.1 M KOH + 3 M CH3OH solutions, respectively. No change of the current track of ORR in both solutions is observed for Fe-N-CNT@GN from Fig. 7a. For the Pt/C catalyst, however, a sharp change (Fig. 7b) in the current density (converting from negative current for the ORR to positive current for the methanol oxidation reaction at the potential range of −0.3 to 0.3 V) indicates the susceptibility to methanol crossover. This result confirms that the Fe-N-CNT@GN catalyst performs much better tolerance to methanol crossover effect and higher fuel selectivity toward ORR than commercial Pt/C.
 |
| | Fig. 7 (a) CV curves of Fe-N-CNT@GN and (b) Pt/C in O2-saturated 0.1 M KOH and O2-saturated 0.1 M KOH with 3 M CH3OH at a scan rate of 10 mV s−1. | |
4. Conclusions
In summary, we report a facile method to prepare CNTs supported iron, nitrogen elements, which was finally encapsulated with graphene by hydrothermal method. The Fe-N-CNT@GN catalyst exhibits higher catalytic performance for ORR in an alkaline medium, the ORR onset potential of −0.13 V, the half wave potential of −0.25 V and the highest limiting current density of 6.2 mA cm−2. And the Fe-N-CNT@GN catalyst displays superior electrocatalytic stability and methanol tolerance. These indicate that the Fe-N-CNT@GN catalyst can be a promising non-precious metal cathode catalyst for fuel cells. Comprehensively, analyzing the XRD, XPS, and RDE/RRDE results, we deduce that both Fe3N (where Fe ions probably bonded with pyrrolic nitrogen) and graphitic nitrogen incorporated into the carbon matrix are the possible ORR active sites, which enhance the ORR kinetic activity. Graphene layers protect the active sites from the corrosion of electrolyte and enhance the durability of the catalyst.
Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (Grant No. DUT14RC (3)078, DUT16ZD102) and the State Key Laboratory of Fine Chemicals (Panjin) project (Grant No. JH2014009).
Notes and references
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- B. Wang, J. Power Sources, 2005, 152, 1–15 CrossRef CAS.
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 133–144 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- H. W. Liang, X. Zhuang, S. Brüller, X. L. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744 CAS.
- J. C. Meier, I. Katsounaros, C. Galeano, H. J. Bongard, A. A. Topalov, A. Kostka, A. Karschin, F. Schüth and K. J. J. Mayrhofer, Energy Environ. Sci., 2012, 5, 9319 CAS.
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 145–154 CrossRef CAS.
- R. Othman, A. L. Dicks and Z. H. Zhu, Int. J. Hydrogen Energy, 2012, 37, 357–372 CrossRef CAS.
- H. Schulenburg, S. Stankov, V. Schünemamn, J. Radnik, I. Dorbandt, S. Fiechter, P. Bogdanoff and H. Tributsch, J. Phys. Chem. B, 2003, 107, 9034–9041 CrossRef CAS.
- J. Yang and J. J. Xu, Electrochem. Commun., 2003, 5, 306–311 CrossRef CAS.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- S. T. Chang, H. C. Hsu, H. C. Huang, C. H. Wang, H. Y. Du, L. C. Chen, J. F. Lee and K. H. Chen, Int. J. Hydrogen Energy, 2012, 37, 13755–13762 CrossRef CAS.
- S. T. Chang, C. H. Wang, H. Y. Du, H. C. Hsu, C. M. Kang, C. C. Chen, J. C. S. Wu, S. C. Yen, W. F. Huang, L. C. Chen, M. C. Lin and K. H. Chen, Energy Environ. Sci., 2012, 5, 5305–5314 CAS.
- C. H. Wang, H. C. Huang, S. T. Chang, Y. C. Lin and M. F. Huang, RSC Adv., 2014, 4, 4207–4211 RSC.
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137, 4296–4299 CrossRef CAS PubMed.
- Z. S. Wu, L. Chen, J. Z. Liu, K. Parvez, H. W. Liang, J. Shu, H. Sachdev, R. Graf, X. L. Feng and K. Müllen, Adv. Mater., 2014, 26, 1450–1455 CrossRef CAS PubMed.
- J. Ahmed, Y. Yuan, L. H. Zhou and S. Kim, J. Power Sources, 2012, 208, 170–175 CrossRef CAS.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- F. H. B. Lima, M. L. Calegaro and E. A. Ticianelli, Electrochim. Acta, 2007, 52, 3732–3738 CrossRef CAS.
- X. W. Liu, X. F. Sun, Y. X. Huang, G. P. Sheng, K. Zhou, R. J. Zeng, F. Dong, S. G. Wang, A. W. Xu, Z. H. Tong and H. Q. Yu, Water Res., 2010, 44, 5298–5305 CrossRef CAS PubMed.
- M. Mahmoud, T. A. Gad-Allah, K. M. El-Khatib and F. El-Gohary, Bioresour. Technol., 2011, 102, 10459–10464 CrossRef CAS PubMed.
- Y. J. Wang, D. P. Wilkinson and J. J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed.
- J. Deng, P. J. Ren, D. H. Deng and X. H. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- J. Y. Choi, R. S. Hsu and Z. W. Chen, J. Phys. Chem. C, 2010, 114, 8048–8053 CAS.
- J. F. Liu, L. L. Wang, X. M. Sun and X. Q. Zhu, Angew. Chem., Int. Ed., 2010, 49, 3492–3495 CrossRef CAS PubMed.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefèvre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J. P. Dodelet, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC.
- L. Lin, Q. Zhu and A. W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- J. Zhang, D. P. He, H. Su, X. Chen, M. Pan and S. Mu, J. Mater. Chem. A, 2014, 2, 1242–1246 CAS.
- K. L. Ai, Y. L. Liu, C. P. Ruan, L. H. Lu and G. Q. Lu, Adv. Mater, 2013, 25, 998–1003 CrossRef CAS PubMed.
- W. Ding, Z. D. Wei, S. G. Chen, X. Q. Qi, T. Yang, J. S. Hu, D. Wang, L. J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 125, 11971–11975 CrossRef.
- R. Zhou and S. Z. Qiao, Chem. Commun., 2015, 51, 7516–7519 RSC.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. F. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- Y. L. Zhai, C. Z. Zhu, E. Wang and S. J. Dong, Nanoscale, 2014, 6, 2964–2970 RSC.
- R. Z. Zhang, S. J. He, Y. Z. Lu and W. Chen, J. Mater. Chem. A, 2015, 3, 3559–3567 CAS.
- R. L. Liu, D. Q. Wu, X. L. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- D. Zhou, L. Yang, L. Yu, J. Kong, X. Yao, W. Liu, Z. Xu and X. Lu, Nanoscale, 2015, 7, 1501–1509 RSC.
- M. Q. Wang, W. H. Yang, H. H. Wang, C. Chen, Z. Y. Zhou and S. G. Sun, ACS Catal., 2014, 4, 3928–3936 CrossRef CAS.
- C. W. Tsai, M. H. Tu, C. J. Chen, T. F. Hung, R. S. Liu, W. R. Liu, M. Y. Lo, Y. M. Peng, L. Zhang, J. J. Zhang, D. S. Shy and X. K. Xing, RSC Adv., 2011, 1, 1349 RSC.
- S. H. Liu, J. R. Wu, C. J. Pan and B. J. H. Wang, J. Power Sources, 2014, 250, 279–285 CrossRef CAS.
- Z. L. Li, G. L. Li, L. H. Jiang, J. L. Li, G. Q. Sun, C. G. Xia and F. W. Li, Angew. Chem., Int. Ed., 2015, 54, 1494–1498 CrossRef CAS PubMed.
- W. Wei, H. W. Liang, K. Parvez, X. D. Zhuang, X. L. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1570–1574 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XPS results, CV curves in O2-saturated 0.1 M KOH solution at different rotation rates and Koutecky–Levich plots of the comparative catalysts; CV curves in O2-saturated 0.1 M KOH solution and accelerated aging test results of the Fe-N-C@GN and Pt/C catalysts. See DOI: 10.1039/c6ra13045c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Si-Mei Chen and
Yang-Yang Xie
,
Si-Mei Chen and
Yang-Yang Xie