
Reduction of selective polyaromatic nitrotriptycene via an azoxytriptycene intermediate under ambient conditions using a cobalt/cobalt oxide nanocomposite (CoNC)†
Akbar Mohammada,
Veenu Mishraa,
Prakash Chandraa and
Shaikh M. Mobin*ab
aDiscipline of Chemistry, Indian Institute of Technology Indore, Simrol, Indore 452020, M.P., India. E-mail: xray@iiti.ac.in
bCentre for Material Science and Engineering, Indian Institute of Technology Indore, Simrol, Indore 452020, M.P., India
First published on 27th May 2016
Abstract
A cobalt-based nanocomposite (CoNC) has been prepared from a recently reported single source molecular precursor (SSMP) [CoII(hep-H)(H2O)4]SO4 (A) (hep-H = 2-(2-hydroxylethyl)pyridine). The resulting nanocomposite material was characterized by using various physicochemical techniques such as XRD, SEM, EDAX, TEM and XPS spectroscopy. X-ray diffraction patterns show the weakly crystalline nature of the catalyst. This was also confirmed by the SAED pattern obtained from HR-TEM. XPS analysis reveals the formation of metallic cobalt and the cobalt oxide (CoO) nanocomposite. CoNC was employed for the facile catalytic hydrogenation of 2-nitrotriptycene (M1) and 2,6,14-trinitrotriptycene (M2) as model substrates under atmospheric reaction conditions, which otherwise takes place either with RANEY® Nickel, Pd/C or SnCl2/HCl catalysts under harsh conditions. The mechanistic pathway reveals that the reduction of M1 proceeds via the intermediacy of azoxy triptycene (III) and N-hydroxylamine triptycene (IV).
Introduction
Catalysis has revolutionized today’s chemical manufacturing. One such manufacturing process is the reduction of organic molecules by catalytic hydrogenation. In the last few decades, enormous progress in the field of catalytic hydrogenation of aliphatic/aromatic nitro compounds has been witnessed. In this process, the focus was on improving the conversion yields by varying the (i) reaction conditions, (ii) catalyst from metal to non-metal, and (iii) solvents.1 The catalytic transformation of R-NO2 to R-NH2 proceeds via the formation of versatile intermediates, aryl hydroxylamine, azoxy and azo compounds.2 These products and intermediates can be potentially used in biologically active natural products, dyes, pigments, polymers, pharmaceuticals and agro-chemicals.3 In general, catalytic hydrogenation involves the use of noble or expensive metal catalysts such as Au, Pd, Pt, Rh, Ru or RANEY® Nickel.4 Generally, the use of RANEY® Nickel in industrial processes is believed to be effective due to the fact that no side product except water is formed. However, the key disadvantages of RANEY® Nickel in catalytic hydrogenation are tedious conditions, the use of highly flammable molecular hydrogen, high pressure and high cost. On the other hand, supported catalysts for the reduction of nitroarenes with high intrinsic activity suffer from non-selective hydrogenation leading to undesired by-products and difficult purification.5 Therefore, there is a demand for a more selective, faster and appropriate method for the reduction of R-NO2 to R-NH2.In order to control the high cost and unavailability associated with these metal catalysts, much attention has been paid to economical and robust catalysts as an alternative by using nanoparticles of abundant metals such as Co, Fe, Cu or Ni.6 The Co nanoparticles have gained much interest due to their magnetic nature and wide applications in catalysis, dye adsorption, MRI, biotechnology and data storage.1f,6a,7 The construction and employment of magnetic nanoparticles (MNPs) guarantees non-toxicity, easily accessibility and smooth reproducibility.8 Additionally, the movement and specificity of nano-catalysts can be manipulated by shell modifications.9 Not only metallic cobalt but cobalt oxides too have extensive applications in high density magnetic recording, sensors and heterogeneous catalysis. Co and cobalt oxide have an electronic configuration, dn (n = 5 to 9), which is active for catalytic reduction of nitro compounds to amines.10 The oxides with dn configuration induce reduction by transferring an electron from the BH4− to the nitro-compounds once they get adsorbed on the metal surface. According to previous literature reports, a positive charge on the surface of the dn elements, having p-type semiconductor properties, facilitates an interaction between the metal surface and the donor BH4− ions.10b
To the best of our knowledge, transformation of M1 and M2 to their respective amines by the cobalt based nanocomposite (CoNC) has not been investigated under atmospheric reaction conditions using NaBH4 as a reducing agent.
To date, 2-nitrotriptycene (M1) and 2,6,14-trinitrotriptycene (M2) have been transformed to their respective amines only by using either RANEY® Nickel, Pd/C or SnCl2/HCl catalysts under harsh conditions (Scheme 1).11
 | ||
| Scheme 1 Reduction of M1 and M2 to their respective amines (1 and 2) using RANEY® Nickel, Pd/C, or SnCl2 as reported in the literature.11 Conditions: for M1; RANEY® Nickel, N2H4/THF, N2, 60 °C, 90 min. For M2; RANEY® Nickel, N2H4/THF, N2, 60 °C, 6–16 h or Pd/C, N2H4/EtOH, reflux, 12 h or SnCl2·2H2O, HCl/H2O/EtOH, 100 °C, 24 h. | ||
Results and discussion
The present work reports the synthesis and characterization of a novel cobalt/cobalt oxide nanocomposite (CoNC). CoNC was derived from a recently reported monomeric Co(II) complex, [CoII(hep-H)(H2O)4]SO4 (A) (Scheme 2), as a single source molecular precursor (SSMP) by a facile method in aqueous media in the presence of NaBH4 at room temperature (Scheme 2).12 | ||
| Scheme 2 General scheme for the synthesis of the molecular precursor and CoNC. | ||
Physicochemical characterization of CoNC was performed using PXRD, TEM, SEM, EDAX, and XPS.13 The crystallinity and phase purity of the material was determined using powder X-ray diffraction. The X-ray diffraction pattern shows the weakly crystalline to amorphous nature of the catalyst (Fig. S1†).
TEM analysis shows that the primary CoNC is less than 5 nm in diameter. These primary nanoparticles aggregated to form secondary particles with a capsule like structure having sizes of 100–150 nm (Fig. 1A and B). Selected area electron diffraction SAED was performed on samples of the nanoparticle aggregate obtained from HR-TEM which showed a concentric and quite diffused ring pattern indicating the weakly crystalline nature of the material (Fig. 1C). The capsule like structure was further confirmed using SEM images as shown in Fig. S2a–e.†
 | ||
| Fig. 1 TEM images of CoNC; (A) 100 nm magnification; (B) 5 nm magnification; (C) SAED pattern 10 1/nm. | ||
The newly developed CoNC was employed as a catalyst towards the hydrogenation of 2-nitrotriptycene (M1) and 2,6,14-trinitrotriptycene (M2) in the presence of NaBH4 under ambient conditions (Scheme 3). M1 and M2 were prepared by following the previously reported methods.11a Catalytic conversion of M1 was found to proceed via the azoxytriptycene intermediate (III) (see later). No reaction was detected in the absence of a hydrogen source or CoNC. The hydrogenation process was monitored by 1H NMR at regular intervals of 10 min and 5 min for M1 and M2, respectively, at room temperature (Fig. S3 and S4†). Further, the evaluation of the effect of solvent on the hydrogenation process established that dichloromethane (DCM) (Table 1, entry 1, Table S1†) was the best choice, leading to 95% conversion of M1 and M2 to 1 and 2 in 60 and 30 min, respectively. The superior catalytic activity of DCM as compared to ether and ethylacetate was due to its higher polarity and the solubility of the reactant in the following order: DCM > ethyl acetate > ether. To our surprise, moderate conversion of M1/M2 to 1/2 was also observed in water because the lower solubility of the nitrotriptycene played a detrimental role in product selectivity, in spite of the higher polarity of water molecules.
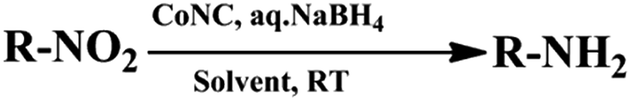 | ||
| Scheme 3 Nitrotriptycene reduction catalysed by NaBH4 and CoNC. | ||
| Entry | Solvent | Catalyst (mol%) (CoNC) | 1 | 2 | ||
|---|---|---|---|---|---|---|
| Yield (%) | Time (min) | Yield (%) | Time (min) | |||
| 1 | DCM | 5 | 95 | 60 | 95 | 30 |
| 2 | EtOAc | 5 | 73 | 70 | 82 | 70 |
| 3 | Ether | 5 | 61 | 70 | 90 | 70 |
| 4 | Water | 5 | 30 | 240 | 66 | 180 |
The comparison of the reaction conditions between the reported RANEY® Nickel, Pd/C or SnCl2 and CoNC as catalysts for the hydrogenation of M1/M2 (Table 2) revealed the superiority of the latter.
| Substrate | Conditions | Catalyst | Yield (%) | Ref. |
|---|---|---|---|---|
| M1 | N2H4, THF, 60 °C, N2, 1.5 h | RANEY® Nickel | 75 | 11a |
| NaBH4, DCM, RT, air, 60 min | CoNC | 95 | This work | |
| M2 | N2H4, THF, 60 °C, N2, 6–16 h | RANEY® Nickel | >90 | 11b–d |
| N2H4, EtOH, reflux, 12 h | Pd/C | 88–90 | 11e–g | |
| HCl, H2O/EtOH, 100 °C 24 h | SnCl2·2H2O | 57 | 11h | |
| NaBH4, DCM, RT, air, 30 min | CoNC | 95 | This work |
The careful monitoring of the reduction of M1 by CoNC via thin layer chromatography (TLC) facilitated the isolation and structural characterization of the intermediate azoxytriptycene (III) which, in turn, extended the mechanistic outlook of the transformation process.
The mechanism of nitro reduction is similar to the proposed mechanism for Cu nanoparticles as reported earlier.14a Like other metals, cobalt is a good conductor of electrical conductivity, facilitating easy electron transfer from one adsorbed species on its surface to another. This property makes CoNC an excellent catalyst for nitro reduction. A probable mechanism for the reduction of nitro compounds by NaBH4 and CoNC has been discussed.
XPS analysis was performed to obtain the elemental composition and chemical and electronic state of CoNC (Fig. 2 and S5E–H†). All the binding energies were calibrated using the C 1s peak at 284.6 eV. The formation of metallic Co nanoparticles was confirmed by the presence of a peak at 778 eV. The presence of peaks at 781.6 eV, 793.2 eV and 787 eV was due to 2p3/2, 2p1/2 and a satellite peak, respectively, and confirms the presence of the Co2+ oxidation state. The aforementioned results show that complex A is reduced to metallic Co and its oxides CoO or Co2O3. The presence of a peak at 532.6 eV in both fresh and spent catalyst confirmed the presence of O 1s (Fig. S5G and H†).14b,c The reduction of complex A with NaBH4 results in the formation of CoNC. Metallic Co nanoparticles, present in CoNC, catalyzed nitrotriptycene reduction according to the classical Langmuir–Hinshelwood model as is depicted in Chart 1A. BH4− and 2-nitrotriptycene sequentially get adsorbed on the CoNC surface. The adsorption of the nitrotriptycene is followed by the transfer of surface-hydrogen species and electrons (reduction) from BH4− to 2-nitrotriptycene on its surface to furnish 2-aminotripticyene.
 | ||
| Fig. 2 XPS spectra; (A) fresh CoNC; (B) Co present in CoNC; (C) spent CoNC; (D) Co present in spent CoNC. | ||
 | ||
| Chart 1 Probable mechanistic pathway for nitrotriptycene reduction using (A) metallic Co nanoparticles; (B) cobalt oxide nanoparticles. | ||
During the catalytic hydrogenation process, the formation of the Co2O3 or Co3O4 composites via partial oxidation of metallic Co nanoparticles in alkaline media was also observed. XPS analysis of the spent catalyst did not show an appearance of any peak at 778 eV, confirming that there was oxidation of the metallic Co nanoparticles to Co2+ during the reaction. Oxidation of cobalt nanoparticles was further confirmed using SEM-EDAX analysis. EDAX of fresh and spent catalyst showed the presence of Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O, in an atomic ratio of 65.59:34.51 and 35.08:64.08 respectively, to further confirm the oxidation of metallic cobalt and cobalt oxide to their higher oxidation states (Fig. S2d and e and S6c and d†). This results in the metal oxide nanocomposite having p-type semiconductor properties. A plausible mechanism for the reduction of M1 to 1 on the metal oxide starts with (i) chemisorption of BH4− on the surface of the metal oxide followed by (ii) discharge of electrons from BH4− through the metal oxide to the acceptor (Chart 1B).
:O, in an atomic ratio of 65.59:34.51 and 35.08:64.08 respectively, to further confirm the oxidation of metallic cobalt and cobalt oxide to their higher oxidation states (Fig. S2d and e and S6c and d†). This results in the metal oxide nanocomposite having p-type semiconductor properties. A plausible mechanism for the reduction of M1 to 1 on the metal oxide starts with (i) chemisorption of BH4− on the surface of the metal oxide followed by (ii) discharge of electrons from BH4− through the metal oxide to the acceptor (Chart 1B).
Water used for dissolving NaBH4 plays the role of a polar protic solvent, giving the required H+ ions necessary for the reduction of the nitro group to amines. Alternatively, it can also be postulated that cobalt oxide nanoparticles were in situ reduced to Co(0) nanoparticles with the help of NaBH4 as a reducing agent. These Co(0) nanoparticles were responsible for the reduction of nitro-aromatics to amines.10c As depicted in Chart 1B, the reduction of M1 to 1 is briefly discussed here. M1 is simultaneously attacked by NaBH4 molecules on the CoNC surface to form a nitrosotriptycene intermediate. During the catalytic process, II is furthermore reduced to hydroxylamine triptycene (IV), and II and IV condense together to form azoxytriptycene (III) (Fig. S7 and S8†). Formation of III was further confirmed using single crystal X-ray structure analysis. The intermediate is further reduced to 1 via multiple hydrogenation steps.
The structure of III (Table S2†)‡established that two units of triptycene were connected by the azoxy group (Chart 1: III, Table S2†)‡. The structure of 2 (Fig. 3 and Table S2†)‡ showed that three (–NH2) groups were positioned at the edges of the three blades of the triptycene unit with interplanar angles of 105.1° between these blades of triptycene. The packing of 2 revealed the presence of intermolecular N–H⋯N and C–H⋯π interactions, leading to a 2D-network (Fig. S9 and Table S3†).
 | ||
| Fig. 3 Crystal structure of 2 (solvent molecule omitted for clarity). | ||
CoNC could be reused for two successive cycles with retention of activity of up to ≈95% but a steep decrease in activity was observed thereafter (Fig. 4). Decrease in the catalytic activity and lifetime of the catalyst during recycling was due to aggregation of cobalt nanoparticles due to their strong magnetic attraction as well as high surface energy amongst them.14a
 | ||
| Fig. 4 Recycling study of CoNC catalysed 2-nirotriptycene reduction using NaBH4 as the reducing agent. | ||
The successful hydrogenation of M1 and M2 by CoNC prompted us to explore the general applicability of the newly adopted process for other polyaromatic nitro compounds (Scheme 4 and Table S1†) using a wide variety of solvents such as DCM, EtOAc, ether, water and MeOH.
 | ||
| Scheme 4 Reduction of other polyaromatic nitro compounds. | ||
Very high conversion and selectivity was obtained for various polyaromatic nitro compounds in varying solvents (ESI Table S1,† entry no. 3–8). The reaction was observed in all studied solvents with higher conversion and good selectivity for entry no 7, except water. In the case of entry no. 8, MeOH and ether gave 99% conversion while the others had either average conversion or poor conversion. Moreover, other substituted polyaromatic compounds with amino, methyl and methoxy functionalities (Table S1,† entry no. 5–6, 8, 10) showed a high conversion (75–99%) and selectivity. However, the product yield for 9 and 10 was slightly lower at 70 and 75%, respectively. However, DCM and MeOH were compatible with almost all of the polyaromatic nitro compounds and the other substituted compounds. High polarity of DCM and MeOH, as well as better solubility of the polyaromatic nitro compounds in these solvents compared to other solvents, was responsible for the superior catalytic performance of CoNC. This facile reduction of compounds was achieved at RT leading to the formation of their respective amines with high conversion in the range of 70–99% (Table S1,† Fig. S10–S17†).
Conclusions
To summarize, CoNC was prepared by reduction of a single source molecular precursor (SSMP), A, using NaBH4 as a reducing agent. CoNC was characterized using several physicochemical techniques and then used for the reduction of M1. Powder XRD spectra indicated the formation of a weakly crystalline to amorphous material. HRTEM images and SAED patterns further corroborated the XRD data. Elemental analysis performed using XPS spectroscopy of CoNC revealed the formation of metallic cobalt and cobalt oxide. The superior catalytic feature of the newly developed CoNC with well defined morphology for the reduction of polyaromatic nitro compounds, including M1 and M2 as compared to the reported RANEY® Nickel, Pd/C and SnCl2 based catalysts, was established. Solvent effect was also investigated for M1, and DCM was found to be best solvent for M1 reduction to 1. Furthermore, the probable outlook of the reaction pathway was established via the structural characterization of the azoxytriptycene intermediate (III). The scope of the catalytic material (CoNC) was tested for other polyaromatic nitro compounds. CoNC with magnetic properties can be considered as a promising synthon for other potential applications.Experimental section
Materials
Commercially available starting materials, CoSO4·7H2O, 2-(2-hydroxyethyl) pyridine (hep-H), sodium borohydride (NaBH4), nitro compounds and reagent grade solvents were used as received. The model substrates (M1 and M2) were synthesized according to previous reported literature. All of the reagents were of analytical grade and used without further purification.Physical characterizations
Powder X-ray diffraction studies were carried out on a Rigaku SmartLab X-ray diffractometer using CuKα radiation (1.54 Å). FE-SEM was carried out with attached EDAX using a Supra55 Zeiss Field-Emission Scanning Electron Microscope. Transmission Electron Microscopy was carried out on a FEI Tecnai G2 12 Twin TEM. XPS analysis of fresh and spent CoNC was performed using X-ray Photoelectron Spectroscopy (XPS) with an Auger Electron Spectroscopy (AES) Module: Model/Supplier: PHI 5000 Versa Probe II, FEI Inc. 1H NMR spectra were recorded on a BrukerAvance(III) 400 MHz spectrometer. Mass analysis was carried on a Brucker-Daltonics, micrOTOF-Q II mass spectrometer. Single-crystal X-ray structural studies were performed on a Supernova Agilent X-ray diffractometer.X-ray crystallographic determination
Single crystal X-ray structural studies of compounds 2 and III were performed on a CCD equipped SUPERNOVA diffractometer from Agilent Technologies with a low-temperature attachment. Crystals of 2 and III were weakly diffracting in nature, and the III molecule shows high disorder, which was refined with model atoms for two positions. Data for 2 and III were collected at 293(2) K using Cu Kα radiation with λα = 1.5418 Å. The strategy for the data collection was evaluated by using the CrysAlisPro CCD software. The data were collected by standard phi-omega scan techniques, and were scaled and reduced using CrysAlisPro RED software. The structures were solved by direct methods using SHELXS-97 and refined by full matrix least squares with SHELXL-97, refining on F2.15 The positions of all of the atoms were obtained by direct methods. All non-hydrogen atoms were refined anisotropically. The remaining hydrogen atoms were placed in geometrically constrained positions and refined with isotropic temperature factors, generally 1.2× Ueq of their parent atoms. All of the H-bonding interactions, mean plane analyses, and molecular drawings were obtained using the program Diamond (ver. 2.1d). The crystal and refinement data are summarized in Table S2† and hydrogen bonding parameters are shown in Table S3.†Preparation of the single source molecular precursor
Co(II) complex, [CoII(hep-H)(H2O)4]SO4 (A), has been synthesized using the previously reported procedure.12Procedure for the synthesis of CoNC
In the typical synthesis of CoNC, 1 mmol of the precursor was dissolved in 20 ml of distilled water and stirred for 5 min. An aqueous solution of NaBH4 was added to the reaction mixture drop-wise until the hydrogen ceased to evolve. The color change from orange to black was observed as the NaBH4 solution was added. Finally the product was washed repeatedly with water and ethanol to remove unreacted materials.Procedure for catalytic reduction
CoNC (5 mol%) was taken in a glass vial, followed by the addition of 5 ml of solvent and 0.1 mmol of nitro compound and aqueous NaBH4 (10 equivalents), and the reaction was magnetically stirred at RT in air for the desired duration of time. The progress of the reaction was monitored by TLC. After completion of the reaction, the product was isolated using normal workup procedures.Acknowledgements
The authors are grateful to CSIR, New Delhi and IIT Indore for funding. We are also grateful to Dr Sampak Samanta for his support and the Advanced Imaging Center, for extending the TEM facility and Advanced Center for Materials Science (ACMS) for XPS, IIT Kanpur. A. M. and V. M. are grateful to MHRD, Government of India for research fellowships and SIC, IIT Indore for providing the characterization facility. V. M. also thanks CSIR, New Delhi for a senior research fellowship.Notes and references
- (a) N. Yan, C. Xiao and Y. Kou, Coord. Chem. Rev., 2010, 254, 1179–1218 CrossRef CAS; (b) J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1–29 CrossRef CAS; (c) N. Yan, Y. Yuan and P. J. Dyson, Dalton Trans., 2013, 42, 13294–13304 RSC; (d) M. Shokouhimehr, J. E. Lee, S. I. Han and T. Hyeon, Chem. Commun., 2013, 49, 4779–4781 RSC; (e) Y. Lin, S. Wu, W. Shi, B. Zhang, J. Wang, Y. A. Kim, M. Endod and D. S. Su, Chem. Commun., 2015, 51, 13086–13089 RSC; (f) V. Udumula, J. H. Tyler, D. A. Davis, H. Wang, M. R. Linford, P. S. Minsonand and D. J. Michaelis, ACS Catal., 2015, 5, 3457–3462 CrossRef CAS.
- (a) C. Yu, B. Liu and L. Hu, J. Org. Chem., 2001, 66, 919–924 CrossRef CAS PubMed; (b) A. Saha and B. Ranu, J. Org. Chem., 2008, 73, 6867–6870 CrossRef CAS PubMed; (c) X.-B. Lou, L. He, Y. Qian, Y.-M. Liu, Y. Cao and K.-N. Fan, Adv. Synth. Catal., 2011, 353, 281–286 CrossRef CAS; (d) X. Liu, S. Ye, H.-Q. Li, Y.-M. Liu, Y. Cao and K.-N. Fan, Catal. Sci. Technol., 2013, 3, 3200–3206 RSC; (e) C. Srilakshmi, H. V. Kumar, K. Praveena, C. Shivakumara and M. M. Nayak, RSC Adv., 2014, 4, 18881–18884 RSC; (f) R. Easterday, O. Sanchez-Felix, Y. Losovyj, M. Pink, B. D. Stein, D. G. Morgan, M. Rakitin, V. Y. Doluda, M. G. Sulman, W. E. Mahmoud, A. A. Al-Ghamdid and L. M. Bronstein, Catal. Sci. Technol., 2015, 5, 1902–1910 RSC.
- (a) D. V. Jawale, E. Gravel, C. Boudet, N. Shah, V. Geertsen, H. Li, I. N. N. Namboothiri and E. Doris, Chem. Commun., 2015, 51, 1739–1742 RSC; (b) S. Fountoulaki, V. Daikopoulou, P. L. Gkizis, I. Tamiolakis, G. S. Armatas and I. N. Lykakis, ACS Catal., 2014, 4, 3504–3511 CrossRef CAS; (c) R. K. Rai, A. Mahata, S. Mukhopadhyay, S. Gupta, P.-Z. Li, K. T. Nguyen, Y. Zhao, B. Pathak and S. K. Singh, Inorg. Chem., 2014, 53, 2904–2909 CrossRef CAS PubMed.
- (a) J. Fang, J. Li, B. Zhang, X. Yuan, H. Asakura, T. Tanaka, K. Teramura, J. Xie and N. Yan, Nanoscale, 2015, 7, 6325–6333 RSC; (b) B. Sheng, L. Hu, T. Yu, X. Cao and H. Gu, RSC Adv., 2012, 2, 5520–5523 RSC; (c) Y.-M. Lu, H.-Z. Zhu, W.-G. Li, B. Hu and S.-H. Yu, J. Mater. Chem. A, 2013, 1, 3783–3788 RSC; (d) R. J. Rahaim Jr and R. E. Maleczka Jr, Synthesis, 2006, 19, 3316–3340 Search PubMed; (e) Y. Lee, S. Jang, C.-W. Cho, J.-S. Bae, S. Park and K. H. Park, J. Nanosci. Nanotechnol., 2013, 11, 7477–7481 CrossRef; (f) C. Antonetti, M. Oubenali, A. M. R. Galletti, P. Serp and G. Vannucci, Appl. Catal., A, 2012, 421–422, 99–107 CrossRef CAS; (g) J. H. Kim, J. H. Park, Y. K. Chung and K. H. Park, Adv. Synth. Catal., 2012, 354, 2412–2418 CrossRef CAS.
- S. Cai, H. Duan, H. Rong, D. Wang, L. Li, W. He and Y. Li, ACS Catal., 2013, 3, 608–612 CrossRef CAS.
- (a) P. Chen, F. Yang, A. Kostka and W. Xia, ACS Catal., 2014, 4, 1478–1486 CrossRef CAS; (b) C. Jiang, Z. Shang and X. Liang, ACS Catal., 2015, 5, 4814–4818 CrossRef CAS; (c) T. Subramanian and K. Pitchuman, ChemCatChem, 2012, 4, 1917–1921 CrossRef CAS; (d) R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7982–7984 RSC.
- (a) J.-F. Soulé, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550–18553 CrossRef PubMed; (b) L. Tang, Y. Cai, G. Yang, Y. Liu, G. Zeng, Y. Zhou, S. Li, J. Wang, S. Zhang, Y. Fang and Y. He, Appl. Surf. Sci., 2014, 314, 746–753 CrossRef CAS; (c) L.-S. Bouchard, M. S. Anwar, G. L. Liu, B. Hann, Z. H. Xie, J. W. Gray, X. Wang, A. Pines and F. F. Chen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4085–4089 CrossRef CAS PubMed; (d) A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- (a) V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Bassett, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed; (b) K. V. S. Ranganath and F. Glorius, Catal. Sci. Technol., 2011, 1, 13–22 RSC; (c) M. B. Gawande, A. K. Rathi, J. Tucek, K. Safarova, N. Bundaleski, O. M. N. D. Teodoro, L. Kvitek, R. S. Varma and R. Zboril, Green Chem., 2014, 16, 4137–4143 RSC.
- (a) M. B. Gawande, P. S. Brancoa and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC; (b) A. Fihri, M. Bouhrara, U. Patil, D. Cha, Y. Saih and V. Polshettiwar, ACS Catal., 2012, 2, 1425–1431 CrossRef CAS; (c) N. Shah, E. Gravel, D. V. Jawale, E. Doris and I. N. N. Namboothiri, ChemCatChem, 2015, 7, 57–61 CrossRef CAS; (d) N. Shah, E. Gravel, D. V. Jawale, E. Doris and I. N. N. Namboothiri, ChemCatChem, 2014, 6, 2201–2205 CrossRef CAS.
- (a) F. A. Westerhaus, R. V. Jagadeesh, G. Wienhöfer, M.-M. Pohl, J. Radnik, A.-E. Surkus, J. Rabeah, K. Junge, H. Junge and M. Nielsen, Nat. Chem., 2013, 5, 537–543 CrossRef CAS PubMed; (b) H. Ma, H. Wang, T. Wu and C. Na, Appl. Catal., B, 2016, 180, 471–479 CrossRef CAS; (c) N. Yan, Z. Zhao, Y. Li, F. Wang, H. Zhong and Q. Chen, Inorg. Chem., 2014, 53, 9073–9079 CrossRef CAS PubMed; (d) X.-W. Wang, K.-L. Wu, K. Liu, W.-Z. Wang, Y.-X. Yue, M.-L. Zhao, J. Cheng, J. Ming, X.-W. Wei and X.-W. Liu, CrystEngComm, 2015, 17, 734–739 RSC; (e) H. Li, J. Liao and T. Zeng, Catal. Sci. Technol., 2014, 4, 681–687 RSC; (f) J. M. Song, S. S. Zhang and S. H. Yu, Small, 2014, 10, 717–724 CrossRef CAS PubMed.
- (a) J. H. Chong and M. J. MacLachlan, Inorg. Chem., 2006, 45, 1442–1444 CrossRef CAS PubMed; (b) C. Zhang and C.-F. Chen, J. Org. Chem., 2006, 71, 6626–6629 CrossRef CAS PubMed; (c) X. Zhu, C.-L. Do-Thanh, C. R. Murdock, K. M. Nelson, C. Tian, S. Brown, S. M. Mahurin, D. M. Jenkins, J. Hu, B. Zhao, H. Liu and S. Dai, ACS Macro Lett., 2013, 2, 660–663 CrossRef CAS; (d) M. Carta, M. Croad, K. Bugler, K. J. Msayiba and N. B. McKeown, Polym. Chem., 2014, 5, 5262–5266 RSC; (e) T. Bura, F. Nastasi, F. Puntoriero, S. Campagna and R. Ziessel, Chem.–Eur. J., 2013, 19, 8900–8912 CrossRef CAS PubMed; (f) H.-H. Chou, H.-H. Shih and C.-H. Cheng, J. Mater. Chem., 2010, 20, 798–805 RSC; (g) Z. Xu, X. Q. Xiong and L. Cheng, Chin. Chem. Lett., 2008, 19, 1127–1130 CrossRef CAS; (h) S. L. Buchwald, T. M. Swager, G. Teverovskiy and M. Su, PCT Int. Appl. WO 2014070888 A1, 2014.
- S. M. Mobin and A. Mohammad, Dalton Trans., 2014, 43, 13032–13040 RSC.
- (a) Y.-B. Cao, X. Zhang, J.-M. Fan, P. Hu, L.-Y. Bai, H.-B. Zhang, F.-L. Yuan and Y.-F. Chen, Cryst. Growth Des., 2011, 11, 472–479 CrossRef CAS; (b) H. P. Singh, S. Sharma, S. K. Sharma and R. K. Sharma, RSC Adv., 2014, 4, 37816–37825 RSC; (c) R. Das, P. Pachfule, R. Banerjee and P. Poddar, Nanoscale, 2012, 4, 591–599 RSC; (d) X. Liang and L. Zhao, RSC Adv., 2012, 2, 5485–5487 RSC.
- (a) A. K. Sasmal, S. Dutta and T. Pal, Dalton Trans., 2016, 45, 3139–3150 RSC; (b) G. Kwak, J. Hwang, J.-Y. Cheon, M. H. Woo, K.-W. Jun, J. Lee and K.-S. Ha, J. Phys. Chem. C, 2013, 117, 1773–1779 CrossRef CAS; (c) M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, Shelx97, Program for Crystal Structure Solution and Refinement, University of Göettingen, Göettingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization images, spectral data (Fig. S10–S17), a video for catalyst separation and crystallographic details can all be found in the supporting information. CCDC 1422762 and 1422763. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra12920j |
‡ Crystal data for 2: C21H19Cl2N3, M = 384.29, monoclinic P21/n, Z = 4, T = 293(2) K, F(000) = 800, a = 12.084(6) Å, b = 9.0350(12) Å, c = 17.401(6) Å, α = γ = 90°, β = 100.28(3)°, V = 1869.3(11) Å3, Dc = 1.366 mg m−3, μ(Cu Kα) = 3.188 mm−1, size = 0.330 × 0.260 × 0.210 mm3, GOF = 1.076, reflections collected/unique, 3830/1132 [R(int) = 0.0352] R1 [I > 2σ(I)] = R1 = 0.0812, wR2 = 0.2345, R indices (all data) R1 = 0.0850, wR2 = 0.2400. CCDC no. 1422762. Crystal data for III: C40H26N2O, M = 550.63, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 1, T = 293(2) K, F(000) = 576, a = 10.6315(7) Å, b = 11.8390(9) Å, c = 12.1481(8) Å, α = 87.279(6)°, β = 68.228(6)°, γ = 86.725(6)°, V = 1417.08(18) Å3, Dc = 1.290 mg m−3, μ(Cu Kα) = 0.602 mm−1, size = 0.210 × 0.170 × 0.130 mm3, GOF = 1.027, reflections collected/unique, 9377/5357 [R(int) = 0.0223] R1 [I > 2σ(I)] = R1 = 0.0527, wR2 = 0.1382, R indices (all data) R1 = 0.0699, wR2 = 0.1545. CCDC no. 1422763. , Z = 1, T = 293(2) K, F(000) = 576, a = 10.6315(7) Å, b = 11.8390(9) Å, c = 12.1481(8) Å, α = 87.279(6)°, β = 68.228(6)°, γ = 86.725(6)°, V = 1417.08(18) Å3, Dc = 1.290 mg m−3, μ(Cu Kα) = 0.602 mm−1, size = 0.210 × 0.170 × 0.130 mm3, GOF = 1.027, reflections collected/unique, 9377/5357 [R(int) = 0.0223] R1 [I > 2σ(I)] = R1 = 0.0527, wR2 = 0.1382, R indices (all data) R1 = 0.0699, wR2 = 0.1545. CCDC no. 1422763. |
| This journal is © The Royal Society of Chemistry 2016 |