
Ionic liquid binary mixtures: how different factors contribute to determine their effect on the reactivity†
Salvatore Marulloa,
Francesca D'Anna*a,
Paola R. Campodonicob and
Renato Notoa
aDipartimento STEBICEF-Sezione di Chimica, Università degli Studi di Palermo, Palermo, Italy. E-mail: francesca.danna@unipa.it
bCentro de Quimica Medica, Facultad de Medicina, Clìnica Alemana, Universidad del Desarrollo, Santiago, Chile
First published on 14th September 2016
Abstract
We studied how mixing ionic liquids affected the rate of the Diels Alder reaction between 9-anthracenemethanol and N-ethylmaleimide. The mixtures considered spanned the whole compositional range and differed for the anion or the cation. These comprised mixtures of the ionic liquid 1-benzyl-3-butyl-imidazolium bis-(trifluoromethanesulfonyl)imide ([Bzbim][NTf2]) with 1-(2,3,4,5,6-pentafluorophenyl)-3-butyl-imidazolium bis-(trifluoromethanesulfonyl)imide ([Bz(F5)bim][NTf2]) or 1-benzyl-3-butyl-imidazolium tetrafluoroborate ([Bzbim][BF4]). Moreover we studied the reaction in a set of mixtures containing an aliphatic and an aromatic cation, namely [Bzbim+] and N-butyltriethylammonium ([bEt3N+]) sharing the [NTf2−] anion. In this latter case we carried out RLS measurements, UV-Vis spectra of the solvatochromic probe Nile Red and 1H NMR measurements to gain insights into how the structural organization of the mixtures changes as a function of composition. Altogether, these results show a concomitant effect of solvent viscosity and structural organization on the rate of the process. Finally, no obvious correlation was found between the kinetic trend and the polarity of the mixtures, as expressed by solvatochromic parameters.
Introduction
The study of the properties and uses of ionic liquids (ILs) is a standing feature in present-day chemical research, which has expanded to span a plethora of application fields.1 Compared with classical organic solvents, ILs stand out as more environmentally benign for their low flammability and vapour pressure. More importantly, their ionic nature provides chemists with unique reaction media, characterized by a low solvating power, which in several cases can enhance reaction rates or enable new reactive pathways.2,3 This is often coupled with high chemical and thermal stabilities as well as wide electrochemical windows. ILs composed of aromatic anions or cations also exhibit a marked nanostructure,4 which makes them valuable tools for the synthesis of functional materials or nanoparticles with controlled size range.5,6Furthermore, several reports have underlined the importance of these structural features on their behavior as solvents or catalysts.7 The possibility of changing solvent properties by simply varying the nature of the cation or the anion has earned ionic liquids the title of “designer solvents”. Indeed, given the huge number of possible combinations, this approach makes available a very large range of physico-chemical properties. In this context, Seddon proposed mixing different ILs as a means to further enlarge the gamut of properties obtainable.8 Under this view, unlike conventional organic solvents, changing the composition of a mixture of ILs can, in principle, give rise to a “brand-new” ionic solvent, endowed with different characteristics and behavior. This has prompted in the last years the investigation of the physico-chemical effects arising from mixing ILs. The first investigations mainly focused on how bulk physical properties like density, viscosity and molar volume change upon mixing. In particular these studies were aimed at unraveling whether ILs mixtures display ideal behavior. In general ionic liquids mixtures were described to follow a close to ideal behavior with small deviations.9–12 In other words, this picture considers mixtures of ILs as simple mixtures of two liquids, with fairly predictable behavior, even though a complete generalization could not be made.13–15 In fact, small deviations from ideality in volumetric properties upon mixing have been consistently found. These appeared to be more significant when the difference in size between the anions or the alkyl chain lengths of the cations constituting the mixtures were larger.16–18 A different picture emerges from spectroscopic investigations. Notably, Quitevis et al. showed in their elegant study that the nanostructural organization of ILs mixtures changes markedly depended on the difference in size in the anions.19 Finally, computational investigations suggest an even more articulate picture: a close to ideal behavior for bulk physical properties and, at the same time, non-ideality for other parameters depending on ionic association.20 This kind of dichotomy is evident from more recent studies. On the one hand, Welton et al. reported that although non-ideality arises in limited cases, mixtures of ionic liquids could be treated as ideal solutions, allowing for an easy tunability of the mixture properties.15 On the other hand, Rogers et al. described ILs mixtures as double salts18 pointing out that once ionic liquids are mixed, their constituent ions behave independently, giving rise to a new ionic mixture with distinct properties, not necessarily traceable back to the parent salts. Moreover, they found a unique nanoscale structuring, markedly different from that of the parent ILs upon mixing anions with complementary hydrogen bonding donating abilities.21 Notably, a pronounced tendency towards non-ideal mixing has also been observed for protic ILs.22,23 It is worth mentioning that recent studies pointed out that mixing ILs can also give rise to gel phases that retain and in some cases improve the properties of their constituents, thus proving quite interesting from an applicative point of view.24,25 As already said, most of the studies reported in literature so far have focused on their physico-chemical properties. Much less investigated is the way in which mixing ILs can influence their solvent behavior, with particular regard to organic reactions occurring in their solutions. In this context, we investigated the behavior of mixtures of ILs with a common cation or anion, by means of spectroscopic measurements and studying the kinetics of a probe reaction, namely the piperidine-catalyzed rearrangement of the Z-phenylhydrazone of 3-benzoyl-5-phenyl-1,2,4-oxadiazole into the relevant 1,2,3-triazole.26 In agreement with previous reports, we found that the behavior of the mixtures is mainly governed by the difference in size of the anions. Moreover the extent to which solvent properties change upon mixing has allowed us to use ILs mixtures as suitable reaction media for the conversion of carbohydrates into the chemical platform 5-hydroxymethylfurfural.27
In the attempt to find if any generalization might be possible on the effects exerted by mixing ILs on the solvent behavior of the resulting mixtures, we have carried out an investigation on three sets of ILs mixtures, over the whole compositional range. The mixtures are namely [Bzbim]x[Bz(F5)bim]1−x[NTf2], [Bzbim][BF4]x[NTf2]1−x, and [Bzbim]x[bEt3N]1−x[NTf2] (Chart 1).
 | ||
| Chart 1 ILs mixtures considered and reaction studied. | ||
According to this nomenclature, x is the molar ratio between the ion chosen as reference and the common ion, and coincides with the molar fraction of [Bzbim][NTf2] in the mixture.
In particular, the set of mixtures [Bzbim]x[Bz(F5)bim]1−x[NTf2] is constituted by the same anion and two aromatic cations, while [Bzbim][NTf2]x[BF4]1−x features the same aromatic cation and two anions. Moreover, in [Bzbim]x[bEt3N]1−x[NTf2] aliphatic and aromatic cations share the same anion. We carried out a kinetic study of the Diels Alder reaction between N-ethylmaleimide and 9-anthracenemethanol in each mixture. The mechanism of this reaction is well-understood and proceeds through a cyclic transition state. Consequently this reaction can be a valuable tool to evaluate solvent effects, and has been used in water,28 fluorous solvents29 as well as ionic liquids.30,31 Kinetic measurements were carried out spectrophotometrically, at a temperature of 313 K. For the [Bzbim]x[bEt3N]1−x[NTf2]mixtures, we also carried out a structural study by means of resonance light scattering measurements (RLS) and UV-Vis spectra of the solvatochromic probe Nile Red. RLS provides a means for a qualitative comparison of the size of the aggregates formed since RLS intensity (IRLS) is correlated to this parameter.32 Consequently RLS has been used to probe aggregation in several supramolecular systems, including ILs.26,33–35 On the other hand, Nile Red is a well-established spectroscopic probe, whose absorption and fluorescence are markedly sensitive to the microenvironment in which it is embedded.36–38 To assess whether potential effects deriving from solvent polarity are operative, we determined in this set of mixtures the Kamlet–Taft and ENR polarity parameters. Finally, we recorded 1H NMR of the mixtures to gain insights on the arrangement of cations as a function of composition. Comparing the whole of results obtained in this work with the analogous ones found for a heterocyclic rearrangement reaction,26 could allow us to ascertain whether any generalization on the solvent behavior of ILs mixtures can be drawn or, in contrast, mixing ILs gives rise to articulate, hard to predict outcomes, similarly to what is often observed with pure ILs.39
Results and discussion
In general, studying the same reaction in the three sets of mixtures considered, evidenced very different results. In Fig. 1 we report the plots of the observed rate constant (kobs) obtained in all the mixtures considered, while values of kobs are reported in Tables S1–3.† In all cases, rates change in a non-monotonic fashion as a function of the composition of the mixtures.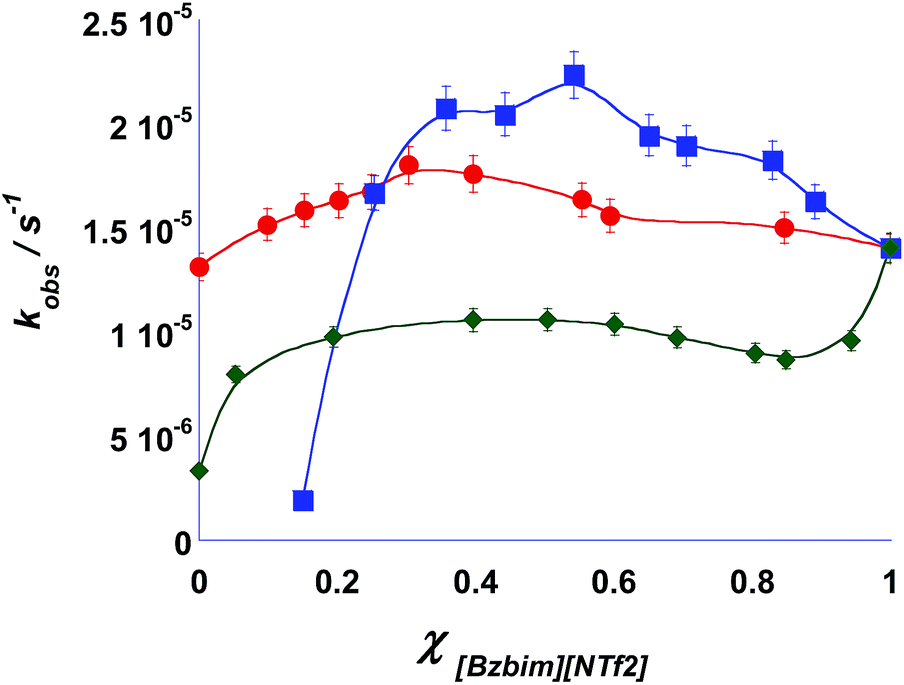 | ||
| Fig. 1 Plots of observed kinetic constant for the Diels–Alder reaction studied in the mixtures (■) [Bzbim][NTf2]x[BF4]1−x, (♦) [Bzbim]x[Bz(F5)bim]1−x[NTf2], (●) [Bzbim]x[bEt3N]1−x[NTf2] at 313.1 K. Lines are displayed only as visual guides. | ||
Some years ago, we obtained a similar trend studying the mononuclear rearrangement of heterocycles in some ILs–organic solvent binary mixtures.40 In that case, the mole fraction corresponding to the highest reactivity was affected by the organic solvent ability to act on the distribution of ion pairs in the IL.
In the present work, when reactions were conducted in the [Bzbim][BF4]x[NTf2]1−x mixtures, we found a bell shaped trend for the observed rate constant (Fig. 1). [Bzbim][NTf2]0.15[BF4]0.85 was the mixture closest to pure [Bzbim][BF4] in which we could determine a kinetic constant. At composition richer in [Bzbim][BF4] we were not able to measure a value for the observed kinetic constant with an acceptable degree of uncertainty, due to the very high viscosity of the resulting solution. Upon adding [Bzbim][NTf2] to the solvent medium, the rate constant increased, reaching a maximum value at a composition of [Bzbim][NTf2]0.55[BF4]0.45. Further enriching the solvent with [Bzbim][NTf2] slowed the process down. At the optimal mixture composition, the ratio of the observed rate constants kmax/k[Bzbim][NTf2]0.15[BF4]0.85, where kmax is the observed rate constant at the maximum, show a ten-fold rate enhancement compared to [Bzbim][NTf2]0.15[BF4]0.85. A less pronounced ratio is found when pure [Bzbim][NTf2] is taken into account (kmax/k[Bzbim][BF4] = 11.5 and kmax/k[Bzbim][NTf2] = 1.58). Notably, the overall trend holds close similarity to what we obtained by studying the rate of a heterocyclic rearrangement reaction in the same mixtures. In this latter case, kinetic investigations showed non-monotonic trends for the mixtures bearing anions of different sizes, whereas the mixtures prepared from ionic liquids sharing the same anion featured a monotonic trend, although with significant deviations from linearity. Similar trends were provided by RLS measurements and UV-Vis spectra of Nile Red.26 Such findings were in agreement with the results obtained by Quitevis et al. by means of OKE spectra. These showed that IL mixtures with a common cation and anions of comparable size featured a random arrangement of ions, compatible with a statistical mixing. In this case a close to ideal behaviour should be expected. Conversely, when the mixture was constituted by anions of different size, the arrangement of anions is no longer random but characterized by the formation of locally ordered domains. In analogy with block co-polymers, such an arrangement is termed as co-block network and should give rise to a less predictable behavior.
Overall these considerations suggest that the way the Diels–Alder reaction responds to changes in the organization of the solvent is similar to that of the heterocyclic rearrangement reaction. In fact, Diels Alder reactions are known to be slowed down in ILs due to their higher viscosity compared with molecular solvents.41 Although viscosity affects the cycloaddition between 9-anthracenemethanol and N-ethylmaleimide in solution of ILs in a complex way,30 we can reasonably state that rising viscosity in the solvent medium slows the overall process down. In the light of this, and considering that [Bzbim][BF4] has a higher viscosity than [Bzbim][NTf2],26 we can infer that in this case a prominent role exerted by viscosity can be ruled out. In fact, as mixtures become richer and richer in [Bzbim][NTf2], solvent viscosity diminishes and consequently an upward trend should be expected, which is not the case. It is worth remembering that [Bzbim][NTf2] and [Bzbim][BF4] show very different structural organizations as accounted for by RLS measurement previously reported,26 indicating the presence of more extended aggregates in the latter. The changes in the solvent organization originated by mixing [Bzbim][BF4] and [Bzbim][NTf2] were thus explained on the grounds of the co-block network organization of the mixture considered.
We observed a very different picture when the reaction was studied in the [Bzbim]x[Bz(F5)bim]1−x[NTf2] mixtures. The plot reported in Fig. 1 shows indeed a quite articulate trend. Upon adding [Bzbim][NTf2] the rate constant increases, reaching a maximum value at [Bzbim]0.4[Bz(F5)bim]0.6[NTf2]. Then, further addition of this IL slows moderately down the process. Finally, at a composition of [Bzbim]0.8[Bz(F5)bim]0.2[NTf2] a steep increase in rate is detected. Rate constants are comparable with those obtained in the mixtures containing different anions [Bzbim][NTf2]x[BF4]1−x. However, in this case, changes in rate are less pronounced. Indeed, kmax/k[Bz(F5)bim][NTf2] = 3.19 and kmax/k[Bzbim][NTf2] = 0.75, where kmax is the observed rate constant at [Bzbim]0.4[Bz(F5)bim]0.6[NTf2]. This result shows a much different behavior from that expected only on the grounds of the structural organization of the mixture. Indeed, RLS measurements showed a monotonic drop in the size of aggregates, consistent with a random network organization on going from [Bz(F)5bim][NTf2] to [Bzbim][NTf2].26 Thus, for the Diels–Alder reaction, factors other than the organization of ions in the mixture should affect the rate of the process and viscosity appears to play a significant role. A closer analysis of the plot reported in Fig. 2b allows us to identify three different regions. For the sake of simplicity we will examine each one separately. In the first portion of the plot, rate increases as solvent composition ranges from [Bz(F5)bim][NTf2] to [Bzbim]0.4[Bz(F5)bim]0.6[NTf2]. Based on the hypothesis that viscosity and solvent organization act together and independently on the process rate, and bearing in mind that [Bz(F5)bim][NTf2] is more viscous than [Bzbim][NTf2],26 we should expect a positive contribution deriving from the drop in viscosity and a negative one ensuing from the decrease in structural organization. Consequently, in this region the effect of the reduction in viscosity prevails and dictates the observed outcome. In the second region, between [Bzbim]0.4[Bz(F5)bim]0.6[NTf2] and [Bzbim]0.8[Bz(F5)bim]0.2[NTf2], only slight changes in rate are detected. This suggest that in this composition range, viscosity and solvent organization exert effects of comparable entity. Finally in the third region, spanning compositions from [Bzbim]0.8[Bz(F5)bim]0.2[NTf2] to [Bzbim][NTf2], the rate of the process rapidly increases. In agreement with the above picture, the solvent organization in this mixture reaches its minimum and so the reduction in viscosity is the only parameter affecting the outcome, inducing a significant rise in the observed rate constant.
 | ||
| Fig. 2 Plots of (a) resonance light scattering measurements and (b) UV-Vis spectra of Nile Red in the mixtures [Bzbim]x[bEt3N]1−x[NTf2]. | ||
Given this premises, it may seem contradictory that for the same reaction, the rate is governed mainly by solvent organization in the [Bzbim][NTf2]x[BF4]1−x mixtures and also by solvent viscosity in the [Bzbim]x[Bz(F5)bim]1−x[NTf2] ones. This can be explained considering the very different extent of variation of solvent organization in the two cases. Indeed, as revealed by RLS measurements, the changes in solvent organization in the former mixtures are much more pronounced than those observed in the latter ones.26 Consequently, when large differences in organization occur, this parameter can outweigh viscosity in determining reaction rates. On the other hand, less pronounced variations in the nanostructural organization of the mixture allow viscosity to manifest its effect on the rate of the process. This is also in agreement with the lower reactivity ratios determined in the [Bzbim]x[Bz(F5)bim]1−x[NTf2] mixtures compared with [Bzbim][NTf2]x[BF4]1−x.
We observed another different picture when the reaction was studied in the [Bzbim]x[bEt3N]1−x[NTf2] mixtures. A first glance at the plot reported in Fig. 1 points out that the observed rate constants follow a bell shaped trend as a function of the composition of the solvent mixture. Once again, increasing the amount of [Bzbim][NTf2] induces a rise in the rate of the process up to an optimal composition, after which it decreases regularly. In this case, the observed rate constants reach the maximum value at the composition of [Bzbim]0.4[bEt3N]0.6[NTf2]. Considering the very different structure of the mixed cations, to our surprise in these mixtures we found the least pronounced kinetic effect if compared with the previous results. Indeed the reactivity ratios kmax/k[Bzbim][NTf2] and kmax/k[bEt3N][NTf2] amount to 1.24 and 1.36 respectively. Comparing these results with those collected in the mixtures [Bzbim]x[Bz(F5)bim]1−x[NTf2], evidences that changing the molar ratio of two cations with similar size and complementary quadrupole moment42 ([Bzbim] and [Bz(F5)bim]), induces more dramatic kinetic effects than mixing two cations differing mainly by size ([bEt3N] and [Bzbim]).
To gain further information on how the structural organization of this set of mixtures change with composition, we carried out RLS measurements and recorded UV-Vis spectra of solutions of Nile Red in the mixtures considered. Plots relevant to both measurements are reported in Fig. 2.
Both investigations point out a discontinuity in close proximity to [Bzbim]0.45[bEt3N]0.55[NTf2]. In particular, the plot reported in Fig. 2b features two clear changes in slope at [Bzbim]0.4[bEt3N]0.6[NTf2] and [Bzbim]0.6[bEt3N]0.4[NTf2]. This suggests that the probe experiences two significantly different microenvironments. Moreover, RLS measurements reveal the presence of more extended aggregates as the molar fraction of [Bzbim][NTf2] in the solvent medium exceeds 0.45. All these observations indicate that the mixtures become more organized upon increasing the amount of [Bzbim][NTf2]. On the other hand, viscosity raises on going from pure [bEt3N][NTf2] to pure [Bzbim][NTf2] (η = 104 cp (ref. 43) and 239 cp,44 for [bEt3N][NTf2] and [Bzbim][NTf2] respectively). Once again, the trend found for the observed rate constant can be explained on the grounds of the competing effects exerted by viscosity and structural organization of the solvent. Indeed, adding [Bzbim][NTf2] to pure [bEt3N][NTf2] induces an initial increase in the observed rate constant as the reaction medium becomes more organized until reaching the composition [Bzbim]0.4[bEt3N]0.6[NTf2]. Then, further enriching the mixture with [Bzbim][NTf2] increases viscosity to an extent sufficient to induce a slowing down of the reaction. It is important to note though, that in this case the overall changes in reactivity are relatively small, indicating that the two effects are comparable in entity over the whole composition range explored. To support our hypothesis, we also determined the polarity parameter ENR45 and the Kamlet–Taft parameters46 β and π* for each [Bzbim]x[bEt3N]1−x[NTf2] mixture. We were not able to determine the Kamlet–Taft parameter α with acceptable accuracy due to protonation of the solvatochromic probe required. The plots of polarity and Kamlet–Taft parameters as a function of the molar fraction of [Bzbim][NTf2] are reported in Fig. S1.† The Kamlet–Taft parameter β is related to the hydrogen bond accepting ability of the solvent. Examination of the plot reported in Fig. S1a† reveals that this parameter does not change significantly over the composition range explored. This is in agreement with the observation that β in ILs is mainly governed by the anion, which in our case is the constituent whose amount in the mixtures stays constant. Moreover, the π* parameter reflects solvent dipolarity/polarizability and in our case regularly increases upon raising the amount of [Bzbim][NTf2]. This is consistent with the mixtures being enriched with an aromatic constituent. Finally, the ENR parameter decreases on going from [bEt3N][NTf2] to [Bzbim][NTf2] and once again a change in slope can be observed at [Bzbim]0.4[bEt3N]0.6[NTf2]. However, none of these trends reflect that of the observed kinetic constant. Consequently, we can rule out that the observed kinetic outcome is governed by changes in bulk solvent polarity to any significant extent.
To gain further insights on the arrangement of cations in this latter set of mixtures we carried out 1H NMR measurements over the whole composition range. In particular we monitored how the position of signals of the [Bzmim] cation varied as the mixtures are enriched with [Bzbim][NTf2].
The plots of the change in chemical shifts (Δδ) of selected protons as a function of composition are reported in Fig. 3 while the spectra are reported in Fig. S2.†
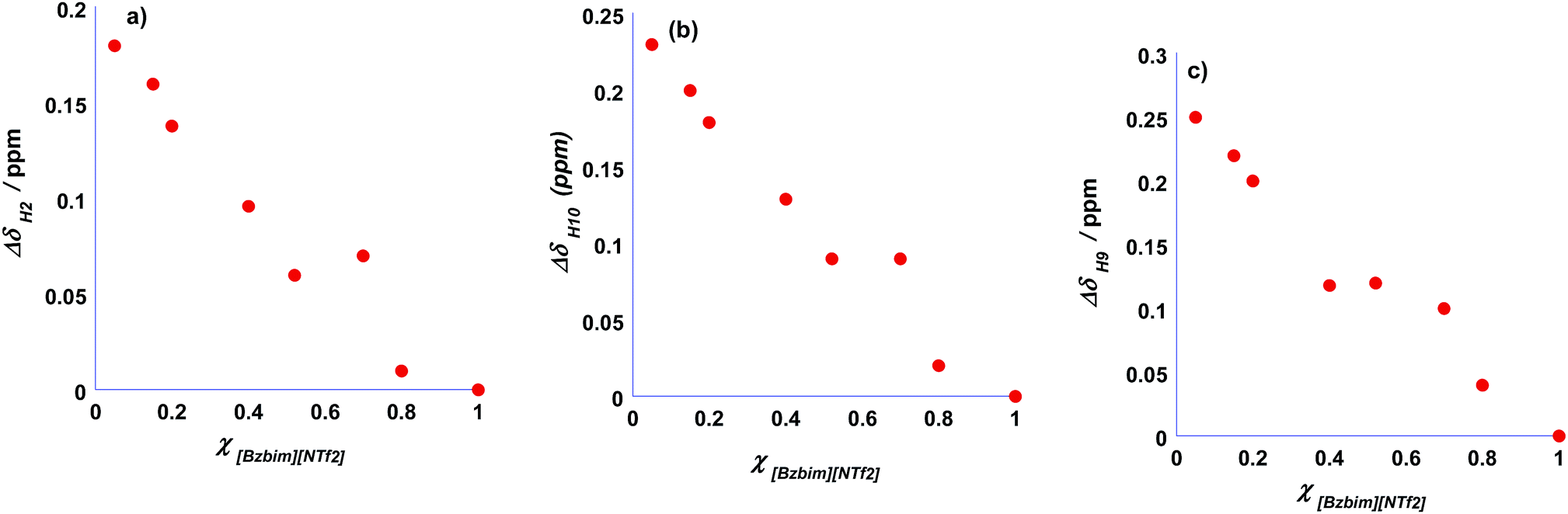 | ||
| Fig. 3 Plots of Δδ as a function χ[Bzbim][NTf2] for (a) H2, (b) H10 and (c) H9 protons. | ||
The signals of H4 and H5 protons in the imidazolium rings are overlapped with those of the phenyl ring, while the rest of aliphatic protons are overlapped with those of the [bEt3N] cation and could not be displayed. In general the signals of H2, H9 and H10 protons in the [Bzbim] cation underwent significant upfield shifts as the mixture composition shifts from a mixture rich in [bEt3N][NTf2] to pure [Bzbim][NTf2]. Variation of chemical shifts ranged from 0.18 for H2 to 0.25 for H9 and H10 protons. It is worth noting that despite the anion is the same along the whole series of mixture, the chemical shift of the H2 protons undergoes a significant upfield shift as the amount of aromatic cation is raised. This can be explained considering that cations in pure [Bzbim][NTf2] adopt a slipped stacked arrangement in which the phenyl rings interact with the imidazolium moieties by means of π–π interactions. Consequently, in solution of mixtures richer in [Bzbim][NTf2] this arrangement induces a shielding of the imidazolium protons by the phenyl ring. On the other hand, when the amount of [bEt3N][NTf2] is raised, this effect is no longer operative and higher chemical shift are observed. Similar considerations can be drawn for the H10 and H9 protons which are located in proximity of the H2 proton. A closer look to the plots reported in Fig. 3 reveals that the chemical shifts of the H2 and H10 protons vary in a fairly linear fashion, while for the H9 proton a clear discontinuity is found at a composition of [Bzbim]0.4[bEt3N]0.6[NTf2]. Notably, at this composition we consistently observed a discontinuity in the structural organization of the mixtures and in the kinetic outcomes. All these observations suggest that the solvent system constituted by the [Bzbim]x[bEt3N]1−x[NTf2] mixtures gradually transitions from a typical aliphatic IL to an IL constituted by aromatic cations as the amount of [Bzbim][NTf2] is raised.

Conclusions
The study of the rate of the Diels–Alder reaction between 9-anthracenemethanol and N-ethylmaleimide in solutions of IL mixtures pointed out an articulate behavior depending on the constituents of the mixtures. In all cases we found a non-monotonic trend for the observed kinetic constants. In general the process rate appears to be affected by solvent structural organization and viscosity. In agreement with our previous study,26 we found the largest changes in reactivity when the reaction was conducted in mixtures constituted of different anions such as [Bzbim][NTf2]x[BF4]1−x. Mixing IL differing for the size of the anions induces structural variations large enough to outweigh the effect of viscosity, and consequently, the kinetic outcome was mainly dictated by the difference in structural organization induced by mixing. When the reaction was conducted in mixtures bearing different cations such as [Bzbim]x[Bz(F5)bim]1−x[NTf2], and [Bzbim]x[bEt3N]1−x[NTf2], we found smaller changes in the solvent structural organization and the influence of viscosity appears of comparable entity.For the [Bzbim]x[bEt3N]1−x[NTf2] mixtures RLS measurements, UV-Vis spectra of Nile Red and 1H NMR spectra of the mixtures pointed out that when an aromatic and an aliphatic cation share the same anion, the properties of the solvent change more gradually although a strictly linear trend could not be found. Finally, comparing the results obtained in this work with those found by us studying a rearrangement reaction26 points out that the solvent behavior of ILs mixtures markedly depends on a the reaction used as a probe, similarly to what happens when pure ILs are used as solvents. Thus, while bulk properties of the mixtures can be foreseen to a significant extent, their behavior as solvents is harder to predict. From this standpoint ILs mixtures behave as IL double salts.18
Experimental
Materials
Commercially available 9-anthracenemethanol, N-ethylmaleimide and Nile Red were used without further purification. [Bzbim][Br] was synthesized according to a reported procedure.26 [Bzbim][NTf2] and [Bzbim][BF4] were prepared by anion metathesis of the corresponding bromides with LiNTf2 and NaBF4 following reported procedures.26,47 [bEt3N][NTf2] was prepared by anion metathesis of the corresponding chloride with LiNTf2 according to a previously reported procedure.48 All ILs gave negative response to the silver nitrate test and were dried in a vacuum line at 70 °C for 2 hours before use, then kept in a desiccator under argon and over calcium chloride. Each ILs mixture was prepared by weighing the proper amount of ILs in a screw-capped vial. To favor mixing, each mixture was vigorously shaken, sonicated for 1 minute, (200 W, 45 kHz), then left to equilibrate overnight before use. In all cases mixtures appeared homogeneous after this treatment.Kinetic measurements
All kinetic measurements were carried out by using a UV/Vis spectrophotometer equipped with a Peltier temperature controller able to keep temperature constant within 0.1 K. The sample for a typical kinetic run was prepared by mixing in a quartz cuvette 200 μL of a solution of 9-anthracenemethanol in the ILs mixture to 200 μL of a N-ethylmaleimide solution in the same mixture. These solutions were previously thermostated at 313.1 K. The concentrations of 9-anthracenemethanol and N-ethylmaleimide were kept constant at 5 × 10−4 M and 4.5 × 10−2 M, respectively. Kinetic runs were carried out by following the disappearance of 9-anthracenemethanol. All ILs mixtures were transparent at the working wavelength.All reactions were followed for at least three half-lives. Apparent kinetic first order constant were reproducible within ±5%. Kinetic data were analyzed by means of KALEIDAGRAPH 4.0 software package.
RLS measurements
Resonance light scattering measurements were carried out using a spectrofluorimeter operating in a synchronous mode with excitation and emission wavelength set at the same value. RLS spectra were recorded from 300 to 650 nm with excitation and emission slit set at 1.5 nm.Nile Red UV-Vis measurements
Nile Red absorbance was determined by injecting into a quartz cuvette (optical path 0.2 cm) 75 μL of a concentrated solution of the spectroscopic probe in acetone. After solvent removal under reduced pressure, 400 μL of the ILs mixture were added.Each sample was kept at a temperature of 298 K for 30 minutes. A suitable working wavelength was chosen based on the difference in UV/Vis spectra of Nile Red in each of the pure components of the mixture. The concentration of Nile Red in each sample was 2.0 × 10−4 M.
Solvent parameter determination
Determination of the solvent parameters was carried out by injecting 75 μL of a concentrated solution of spectroscopic probe in acetone into a quartz cuvette (optical path 0.2 cm). After removal of the solvent, 500 μL of ILs mixture was added. The concentration of the probe was 2.0 × 10−4 M. The solution obtained was thermostated at 298.1 K.1H NMR measurements
NMR spectra were recorded on a 300 MHz instrument. For each measurement, a suitable amount of ILs mixture was placed into a 5 mm NMR tube. A coaxial capillary tube loaded with [D6]DMSO was used as the external lock of the NMR magnetic field/frequency, and its signal at δ = 2.56 ppm was used as the 1H NMR external reference.Acknowledgements
We thank MIUR (FIRB 2010RBFR10BF5V) for financial support. PC thanks project RC-130006 CILIS granted by fondo de innovacion para la competitividad del Ministerio de Economia, Fomento y Turismo de Chile, Fondecyt Grant 1150759.References
- J. P. Hallet and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef PubMed.
- J. Dupont, Acc. Chem. Res., 2011, 44, 1223–1231 CrossRef CAS PubMed.
- J. Dupont and P. A. Z. Suarez, Phys. Chem. Chem. Phys., 2006, 8, 2441–2452 RSC.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- J. Dupont, J. Braz. Chem. Soc., 2004, 15, 341–350 CrossRef CAS.
- C. C. Weber, A. F. Masters and T. Maschmeyer, Green Chem., 2013, 15, 2655–2679 RSC.
- C. D. Hubbard, P. Illner and R. van Eldik, Chem. Soc. Rev., 2011, 40, 272–290 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- A. Stoppa, R. Buchner and G. Hefter, J. Mol. Liq., 2010, 153, 46–51 CrossRef CAS.
- A. Stoppa, J. Hunger and R. Buchner, J. Chem. Eng. Data, 2008, 54, 472–479 CrossRef.
- K. Shimizu, M. Tariq, L. P. N. Rebelo and J. N. C. Lopes, J. Mol. Liq., 2010, 153, 52–56 CrossRef CAS.
- E. T. Fox, J. E. F. Weaver and W. A. Henderson, J. Phys. Chem. C, 2012, 116, 5270–5274 CAS.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- G. J. Maximo, R. J. B. N. Santos, P. Brandão, J. M. S. S. Esperança, M. C. Costa, A. J. A. Meirelles, M. G. Freire and J. A. P. Coutinho, Cryst. Growth Des., 2014, 14, 4270–4277 CAS.
- M. T. Clough, C. R. Crick, J. Grasvik, P. A. Hunt, H. Niedermeyer, T. Welton and O. P. Whitaker, Chem. Sci., 2015, 6, 1101–1114 RSC.
- J. N. Canongia Lopes, T. C. Cordeiro, J. M. S. S. Esperança, H. J. R. Guedes, S. Huq, L. P. N. Rebelo and K. R. Seddon, J. Phys. Chem. B, 2005, 109, 3519–3525 CrossRef PubMed.
- G. Annat, M. Forsyth and D. R. MacFarlane, J. Phys. Chem. B, 2012, 116, 8251–8258 CrossRef CAS PubMed.
- G. Chatel, J. F. B. Pereira, V. Debbeti, H. Wang and R. D. Rogers, Green Chem., 2014, 16, 2051–2083 RSC.
- D. Xiao, J. R. Rajian, S. Li, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. B, 2006, 110, 16174–16178 CrossRef CAS PubMed.
- M. Brussel, M. Brehm, A. S. Pensado, F. Malberg, M. Ramzan, A. Stark and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 13204–13215 RSC.
- H. Wang, S. P. Kelley, J. W. Brantley, G. Chatel, J. Shamshina, J. F. B. Pereira, V. Debbeti, A. S. Myerson and R. D. Rogers, ChemPhysChem, 2015, 16, 993–1002 CrossRef CAS PubMed.
- K. Fumino, A.-M. Bonsa, B. Golub, D. Paschek and R. Ludwig, ChemPhysChem, 2015, 16, 299–304 CrossRef CAS PubMed.
- D. Paschek, B. Golub and R. Ludwig, Phys. Chem. Chem. Phys., 2015, 17, 8431–8440 RSC.
- F. D'Anna, C. Rizzo, P. Vitale, G. Lazzara and R. Noto, Soft Matter, 2014, 10, 9281–9292 RSC.
- P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC.
- F. D'Anna, S. Marullo, P. Vitale and R. Noto, ChemPhysChem, 2012, 13, 1877–1884 CrossRef PubMed.
- F. D'Anna, S. Marullo, P. Vitale, C. Rizzo, P. Lo Meo and R. Noto, Appl. Catal., A, 2014, 482, 287–293 CrossRef.
- A. Kumar and S. S. Pawar, Tetrahedron, 2002, 58, 1745–1749 CrossRef CAS.
- K. E. Myers and K. Kumar, J. Am. Chem. Soc., 2000, 122, 12025–12026 CrossRef CAS.
- S. Tiwari and A. Kumar, J. Phys. Chem. A, 2012, 116, 1191–1198 CrossRef CAS PubMed.
- N. D. Khupse and A. Kumar, J. Phys. Chem. A, 2011, 115, 10211–10217 CrossRef CAS PubMed.
- Y. Cui, F. Cui, L. Wang, Q. Zhang, W. Xue, F. Jing and J. Sun, J. Lumin., 2008, 128, 1719–1724 CrossRef CAS.
- L. P. Jameson and S. V. Dzyuba, RSC Adv., 2013, 3, 4582–4587 RSC.
- B. L. Walton, U. Joshi, S. V. Dzyuba, W. J. Youngblood and G. F. Verbeck, Rapid Commun. Mass Spectrom., 2013, 27, 1954–1960 CrossRef CAS PubMed.
- W. Wang, Y. Zeng, Q. Lu, Y. Xiao, L. Li, Z. Ye and M. Pang, Chin. J. Chem., 2010, 28, 1177–1183 CrossRef CAS.
- R. Vanyúr, L. Biczók and Z. Miskolczy, Colloids Surf., A, 2007, 299, 256–261 CrossRef.
- S. Uppili, K. J. Thomas, E. M. Crompton and V. Ramamurthy, Langmuir, 2000, 16, 265–274 CrossRef CAS.
- L. Di Marzio, C. Marianecci, M. Petrone, F. Rinaldi and M. Carafa, Colloids Surf., B, 2011, 82, 18–24 CrossRef CAS PubMed.
- D. R. MacFarlane and S. A. Forsyth, in Ionic Liquids as Green Solvents, American Chemical Society, 2003, vol. 856, ch. 22, pp. 264–276 Search PubMed.
- F. D'Anna, V. Frenna, S. La Marca, R. Noto, V. Pace and D. Spinelli, Tetrahedron, 2008, 64, 672–680 CrossRef.
- S. Tiwari and A. Kumar, Angew. Chem., Int. Ed., 2006, 45, 4824–4825 CrossRef CAS PubMed.
- J. H. Williams, Acc. Chem. Res., 1993, 26, 593–598 CrossRef CAS.
- C.-P. Lee, J.-D. Peng, D. Velayutham, J. Chang, P.-W. Chen, V. Suryanarayanan and K.-C. Ho, Electrochim. Acta, 2013, 114, 303–308 CrossRef CAS.
- S. S. Moganty, P. S. Chinthamanipeta, V. K. Vendra, S. Krishnan and R. E. Baltus, Chem. Eng. J., 2014, 250, 377–389 CrossRef CAS.
- J. F. Deye, T. A. Berger and A. G. Anderson, Anal. Chem., 1990, 62, 615–622 CrossRef CAS.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192–5200 RSC.
- F. D'Anna, S. La Marca, P. Lo Meo and R. Noto, Chem.–Eur. J., 2009, 15, 7896–7902 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12836j |
| This journal is © The Royal Society of Chemistry 2016 |