
Studies on structural defects in bare, PVP capped and TPPO capped copper oxide nanoparticles by positron annihilation lifetime spectroscopy and their impact on photocatalytic degradation of rhodamine B†
Abstract
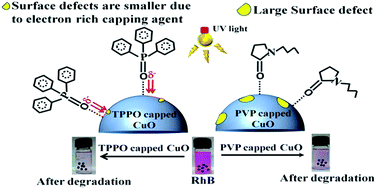
Surface defects are potential sites for modifying the activities of photoexcited electrons and holes, and may consequently affect photocatalytic activity. Here we have demonstrated how surface defects in copper oxide nanoparticles can be engineered by electron rich capping agent (like TPPO) to drastically improve the rate of photocatalytic degradation of rhodamine B dye. The steady state photoluminescence spectroscopy indicated metal vacancies and cluster vacancy defects in the as synthesized batches of bare, PVP (polyvinylpyrrolidone) capped and TPPO (triphenylphosphine oxide) capped copper oxide nanoparticles (NPs). The positron annihilation lifetime spectroscopy studies confirmed that these electron deficient defects were mainly located at the grain boundaries. As compared to the bare and the PVP capped CuO NPs, the positron lifetime parameter ‘τ1’ for TPPO capped CuO NPs was significantly small, which is due to faster annihilation of positrons trapped in free volume defect sites by the local electrons. Unlike PVP, the electron rich TPPO is capable of transporting more electrons to the electron deficient defect sites, due to which the rate of photocatalytic dye degradation was three to four folds higher for the TPPO capped CuO NPs (k = 0.556 h−1) than polyvinylpyrrolidone (PVP) capped CuO NPs (k = 0.168 h−1) and bare CuO NPs (k = 0.143 h−1). The reduced rate of photocatalytic dye degradation for bare and PVP capped CuO NPs were attributable to trapping of photoexcited electrons at the defect sites. The photocatalytic dye degradation was associated with generation of hydroxyl radicals and H2O2 species. The maximum photocatalytic dye degradation by TPPO capped CuO NPs was corroborated with highest concentration of H2O2 detection (125 μM), followed by 79 μM for PVP capped CuO NPs and 65 μM for bare CuO NPs. A detailed mechanism of the role of defects in photocatalytic process has been outlined.
 Please wait while we load your content...
Please wait while we load your content...

