
Structural, kinetic, and DFT studies of the transfer hydrogenation of ketones mediated by (pyrazole)pyridine iron(II) and nickel(II) complexes†‡
Makhosazane N. Magubanea,
George S. Nyamatoa,
Stephen O. Ojwach*a and
Orde Q. Munro*b
aSchool of Chemistry and Physics, University of KwaZulu-Natal, Private Bag X01 Scottsville, 3209, Pietermaritzburg, South Africa. E-mail: ojwach@ukzn.ac.za
bSchool of Chemistry, University of the Witwatersrand, PO WITS 2050, Johannesburg, South Africa. E-mail: orde.munro@wits.ac.za
First published on 29th June 2016
Abstract
A series of iron(II) and nickel(II) complexes chelated by 2-pyrazolyl(methyl)pyridine (L1), 2,6-bis(pyrazolylmethyl)pyridine (L2), and 2,6-bis(pyrazolyl)pyridine (L3) ligands have been investigated as transfer hydrogenation (TH) catalysts for a range of ketones. Nine chelates in total were studied: [Ni(L1)Br2] (1), [Ni(L1)Cl2] (2), [Fe(L1)Br2] (3), [Ni(L2)Br2] (4), [Ni(L2)Br2] (5), [Fe(L2)Cl2] (6), [Ni(L3)Br2] (7), [Ni(L3)Br2] (8), and [Fe(L3)Cl2] (9). Attempted crystallization of complexes 4 and 6 afforded stable six-coordinate cationic species 4a and 6a with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand:metal (L:M) stoichiometry, as opposed to the monochelates that function as precursors to catalytic species for TH reactions. Crystallization of 7·4H2O and 8·2H2O, in contrast, afforded tri- and bis(aqua) salts of L3 chelated to Ni(II) in a 1:1 L:M stoichiometry, respectively. Complexes 1–9 formed active catalysts for the TH of a range of ketones in 2-propanol at 82 °C. Both the nature of the metal ion and ligand moiety had a discernible impact on the catalytic activities of the complexes, with nickel(II) chelate 5 affording the most active catalyst (kobs, 4.3 × 10−5 s−1) when the inductive phase lag was appropriately modelled in the kinetics. Iron(II) complex 3 formed the most active TH catalyst without a significant inductive phase lag in the kinetics. DFT and solid angle calculations were used to rationalize the kinetic data: both steric shielding of the metal ion and electronic effects correlating with the metal–ligand distances appear to be significant factors underpinning the reactivity of 1–9. Catalysts derived from 1 and 9 exhibit a distinct preference for aryl ketone substrates, suggesting the possible involvement of π-type catalyst⋯substrate adducts in their catalytic cycles. A catalytic cycle involving only 4 steps (after induction) with stable DFT-simulated structures is proposed which accounts for the experimental data for the system.
:1 ligand:metal (L:M) stoichiometry, as opposed to the monochelates that function as precursors to catalytic species for TH reactions. Crystallization of 7·4H2O and 8·2H2O, in contrast, afforded tri- and bis(aqua) salts of L3 chelated to Ni(II) in a 1:1 L:M stoichiometry, respectively. Complexes 1–9 formed active catalysts for the TH of a range of ketones in 2-propanol at 82 °C. Both the nature of the metal ion and ligand moiety had a discernible impact on the catalytic activities of the complexes, with nickel(II) chelate 5 affording the most active catalyst (kobs, 4.3 × 10−5 s−1) when the inductive phase lag was appropriately modelled in the kinetics. Iron(II) complex 3 formed the most active TH catalyst without a significant inductive phase lag in the kinetics. DFT and solid angle calculations were used to rationalize the kinetic data: both steric shielding of the metal ion and electronic effects correlating with the metal–ligand distances appear to be significant factors underpinning the reactivity of 1–9. Catalysts derived from 1 and 9 exhibit a distinct preference for aryl ketone substrates, suggesting the possible involvement of π-type catalyst⋯substrate adducts in their catalytic cycles. A catalytic cycle involving only 4 steps (after induction) with stable DFT-simulated structures is proposed which accounts for the experimental data for the system.
Introduction
Transfer hydrogenation (TH) of ketones is one of the most widely used processes for the reduction of ketones to alcohols due to its high selectivity.1 Hydrogenation reactions are mostly applied in industry for the synthesis of fine chemicals, in the pharmaceutical industry for the synthesis of certain alcohol compounds, in the synthesis of agrochemicals, as well as the production of fragrances and flavours.2–4 This process is usually catalysed by homogeneous transition-metal complexes in the presence of a base and an alcohol (typically 2-propanol) as the source of hydrogen. The reaction may be catalysed non-stereoselectively (Scheme 1) with an achiral transition metal complex (TH) or stereoselectively using a chiral catalyst, in which case it is referred to as asymmetric transfer hydrogenation (ATH). To date, the most common metal complexes that have been investigated and found to be active catalysts for the reduction of ketones are ruthenium based.5 Notably, several Ru(II) complexes are active TH catalysts for ketones,6 including complexes of the planar tridentate NNN-donor ligands 2-[6-(3,5-dimethyl-1H-pyrazol-1-yl)pyridin-2-yl]-1H-benzimid-az-ole7 and 2,6-bis(3,5-dimethyl-pyrazol-1-yl)-pyrid-ine.8,9 Ruthenium, however, is expensive and therefore not economically suitable for industrial applications. It is also environmentally unfriendly and is toxic in its ionic form.10 The development of iron11 and nickel12 catalysts for the transfer hydrogenation of ketones potentially offers some advantages over the ruthenium catalysts. For instance, both nickel and iron are relatively cheap, more abundant, and environmentally benign compared with ruthenium.13 Current efforts in the field recognize these attributes and Meyer et al.14 have reported that iron(II) compounds of ethylenediamine-derived diimino-diphosphine ligands form effective catalysts in the transfer hydrogenation of ketones. Homogeneous Ni(0) carbene complexes,15 Ni(0)-based nanoparticles,16,17 and even NiBr2 in a binary NaOH–iPrOH mixture18 are evidently also potentially useful catalysts for the TH of ketones. Recently, chiral Ni(II) complexes chelated by PNO-donor ligands have been used as catalysis in the ATH of a series of aromatic ketones using 2-propanol as the hydrogen source.19 Collectively, however, there are comparatively few reports on transfer hydrogenations of ketones in alcohols in the presence of nickel catalysts.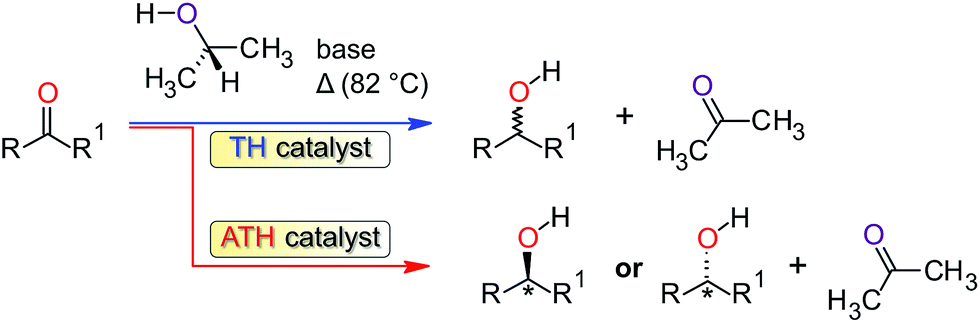 | ||
| Scheme 1 Catalytic transfer hydrogenation (TH) of ketones using 2-propanol. | ||
As part of our continued investigation of late transition metal-catalysed TH reactions of ketones,20 we herein report the use of iron(II) and nickel(II) complexes of three (pyrazolyl)pyridine-based ligands as TH catalysts for a range of ketone substrates (compounds 1–9, Scheme 2). Since TH catalysts for ketones typically operate via an induction phase (activation of a precatalyst to an active catalytic species), they are intrinsically difficult to delineate mechanistically, especially when paramagnetic intermediates are involved. We have accordingly attempted to understand some of the structural and electronic factors of the precatalysts that potentially impact on the reactivity of 1–9 using a combination of DFT simulations and solid angle calculations. By combining the information from the experimental kinetics and insights gained from DFT simulations we have proposed a 4-step catalytic cycle (after induction) that accounts for the data at hand.
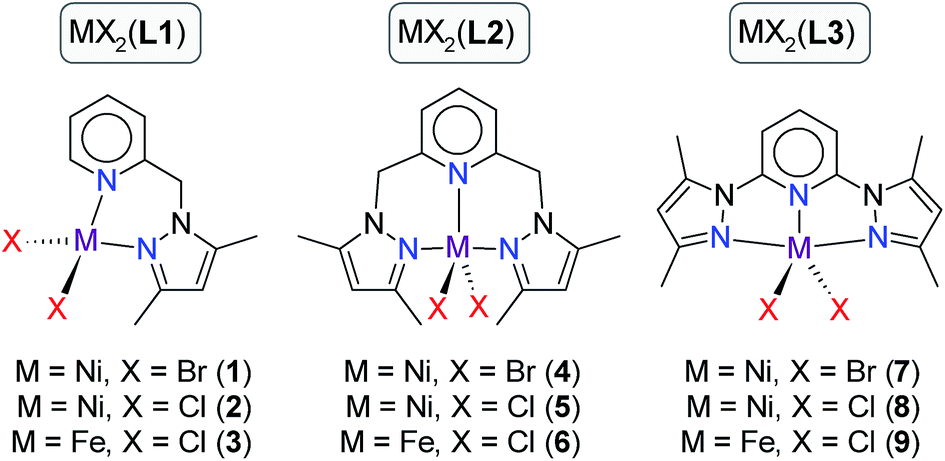 | ||
| Scheme 2 Iron(II) and nickel(II) complexes of (pyrazole)pyridine-based ligands L1–L3 used as precatalysts for the TH of ketones. | ||
Results and discussion
Synthesis of Fe(II) and Ni(II) complexes
The ligands 2-(pyrazolylmethyl)pyridine (L1), 2,6-bis-(pyrazolyl-methyl)-pyridine (L2), and 2,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)pyridine (L3) and their iron(II) and nickel(II) complexes 1–9, were synthesized following previously reported procedures in good yields (61–95%).21 A combination of microanalyses, magnetic moment determinations, and mass spectroscopy were used to confirm the structures of paramagnetic complexes 1–9. The effective magnetic moments of the Ni(II) and Fe(II) complexes were in the range of 2.89–3.79 BM and 4.98–5.01 BM, respectively, consistent with high-spin Ni(II) and Fe(II).22 Moreover, analysis of the mass spectral fragmentation patterns of 1–9 confirmed the dihalide structures depicted in Scheme 2. For instance, the M+ ion of 5 fragments into several diagnostic moieties (Fig. S1†): the base peak at m/z = 188 reflects a 2-pyrazolyl-(methyl)pyridine fragment derived from the parent ligand (L2). The peaks at m/z = 355 (M+ − 2Cl; 20% relative abundance) and m/z = 340 (M+ − 2Cl − CH3; 18% relative abundance) correspond to two stable high molecular weight fragments of 5. Finally, the highest molecular weight fragment at m/z = 386 (M − Cl− – H+) is consistent with the largest intact net anionic fragment, M−, derived from 5 by loss of one chloride ligand and two protons (effectively loss of HCl and H+).X-ray crystal structures
Attempted crystallization of 4 and 6 afforded only the 2:1 ligand:metal adducts 4a and 6a as a result of ligand exchange during re-crystallization in dichloromethane/hexane mixtures. As neither 4a nor 6a were used as TH catalyst precursors, their X-ray structures are given in the ESI (Fig. S2–S5†). The stability of these six-coordinate Ni(II) and Fe(II) complexes is of possible significance to the mechanism of catalyst deactivation we encountered during TH reactions (vide infra).
Fig. 1 summarizes the key X-ray data for 7 and 8, whose structures are similar in several respects, particularly insofar as they both exhibit aquation of metal ion with varying degrees of halide ion substitution; they are hence best classified as inner-sphere hydrates. The Ni(II) ion is six-coordinate in both cases and coordinated to tridentate L3, which deviates somewhat from planarity in both cations. The largest two absolute perpendicular out-of-plane atomic deviations for L3 in complex 7·4H2O measure 0.217 Å (C13) and 0.146 Å (C12), while the root mean square deviation (RMSD) for all non-H atoms is 0.098 Å. In the case of 8, these values are 0.179 Å (C12) and 0.162 Å (Ni1), with a RMSD of 0.092 Å. The origin of the nonplanar structure of L3 in 8 may be traced primarily to a short nonbonded intermolecular C⋯C contact of 3.284(2) Å between C14 of one molecule (a pyrazole carbon) and C8 (the pyridine γ-carbon) of the closest neighbour in the lattice (Fig. S6†), i.e. crystal packing effects as opposed to intramolecular steric strain within the chelating ligand.
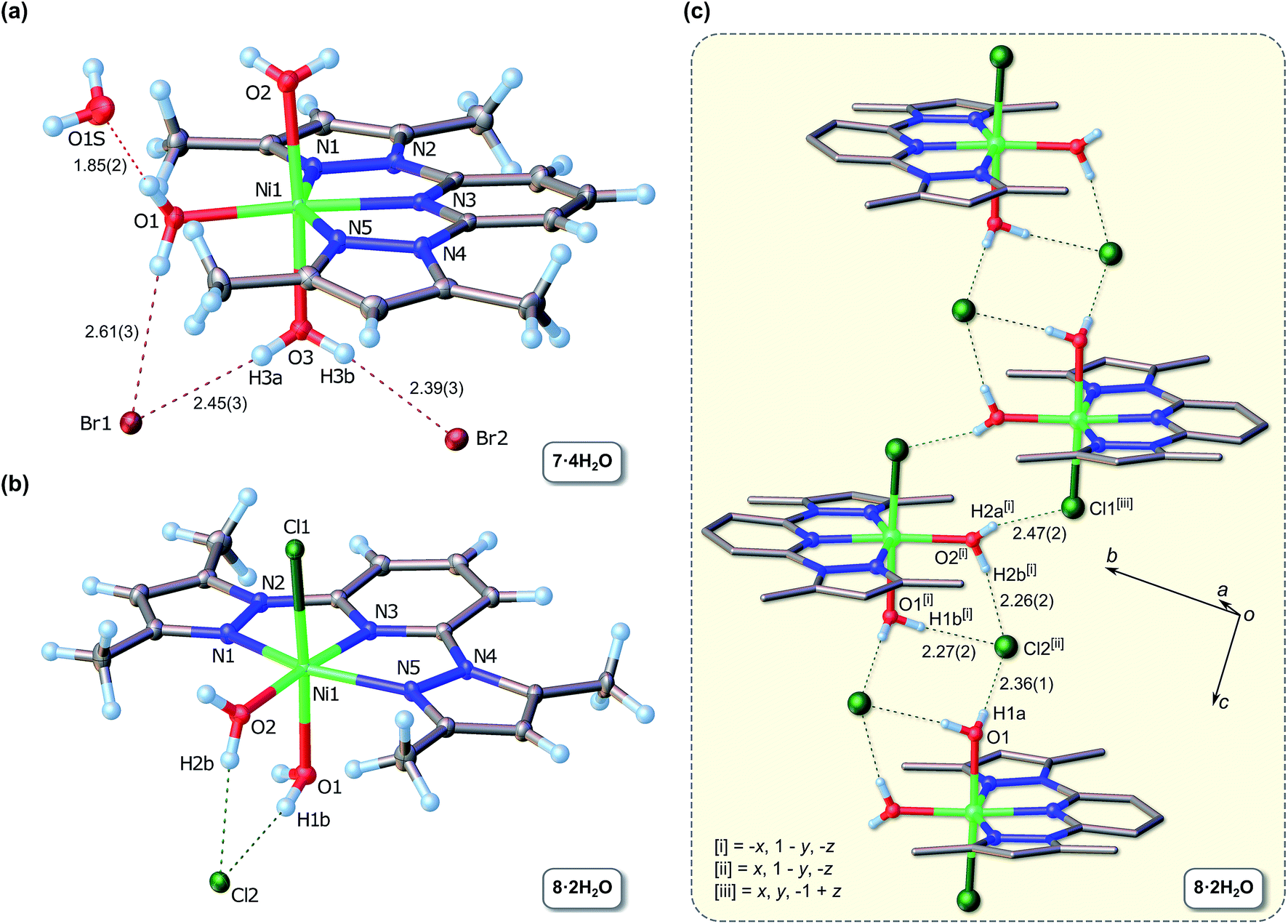 | ||
| Fig. 1 (a) Molecular structure of 7·4H2O drawn with 60% probability ellipsoids, selected atom labels, arbitrary H atom radii, and hydrogen bonds (dashed lines). Bond lengths (Å): Ni1–N1, 2.083(2); Ni1–N5, 2.078(2); Ni1–N3, 2.005(2); Ni1–O1, 2.041(2); Ni1–O2, 2.113(2); Ni1–O3, 2.079(2). Bond angles (°):N5–Ni1–N3, 77.95(7); N3–Ni1–N1, 77.58(7); N1–Ni1–O1, 103.18(7); N5–Ni1–O1, 101.26(7); N5–Ni1–O3, 88.40(7); O3–Ni1–N3, 89.31(7); O3–Ni1–N1, 91.41(7); O3–Ni1–O1, 87.34(7); O1–Ni1–O2, 89.22(6); N3–Ni1–O2, 94.13(7); O2–Ni1–N1, 88.82(7); N5–Ni1–O2, 92.83(7). (b) Molecular structure of 8·2H2O drawn with 50% probability ellipsoids, selected atom labels, arbitrary H atom radii, and hydrogen bonds (dashed lines). Bond lengths (Å): Ni1–N1, 2.0567(8); Ni1–N5, 2.0995(9); Ni1–N3, 2.0162(9); Ni1–O1, 2.0553(9); Ni1–O2, 2.130(1); Ni1–Cl1, 2.4013(4). Bond angles (°):O1–Ni1–N1, 103.78(4); N1–Ni1–N3, 77.61(4); N3–Ni1–N5, 77.02(3); N5–Ni1–O1, 101.32(3); N3–Ni1–Cl1, 94.31(3); N1–Ni1–Cl1, 89.80(3); O1–Ni1–O2, 87.87(3); O2–Ni1–N5, 87.97(3); N5–Ni1–O2, 87.97(3); O2–Ni1–N3, 88.04(3); N1–Ni1–O2, 88.46(3). (c) Illustration of the one-dimensional H-bonded chain formed in 8·2H2O. Selected symmetry-unique H-bond distances are indicated along with relevant symmetry codes. | ||
The bromide salt 7·4H2O exhibits a maximally hydrated coordination sphere with three water ligands occupying coordination sites in the meridional plane perpendicular to the mean plane defined by L3. An additional solvate water molecule is hydrogen-bonded to the aqua ligand bound to Ni1 within the plane of the three N-donors of L3. The bromide counterions in 7·4H2O participate in discrete hydrogen bonds with both the metal-bound and the free water molecules, leading to a complex extended structure (Table S2†). In the structure of 8·2H2O, the two water molecules are bound to the Ni(II) ion both axially and equatorially and are hydrogen-bonded to the chloride counterion. The extended structure comprises one-dimensional H-bonded chains stabilized by centrosymmetric cation and anion pairs (Fig. 1c). The cation pairs are held together by a pair of hydrogen bonds involving the water molecule of one cation and the axial chloride ligand of the partner cation, forming (in graph set notation)23 an 8-membered 2-donor/2-acceptor ring, R22(8). The centro-symmetric anion pair linking adjacent centrosymmetric cation dimers in the chain is characterized by the anions each accepting three hydrogen bonds from the metal-bound water ligands in neighbouring cations to form an 8-membered 4-donor/2-acceptor ring, R24(8), between the cation pairs in the one-dimensional chain. The Ni–O–H⋯Cl− hydrogen bond distances are in the range 2.26–2.47 Å and are consistent with the range (2.10–2.46 Å) determined from 1013 X-ray structures of non-coordinated OW–H⋯Cl− hydrogen bond distances in Steiner's analysis of hydrogen bonding,24 suggesting that the O–H donors of the aqua ligands in 8·2H2O are relatively unaffected by coordination to Ni(II).
The chelating bis(pyrazolyl) ligand in 7 and 8 induces an in-plane distortion of the Ni(II) coordination sphere away from ideal octahedral coordination mainly due to the fact that the Ni(II) ion fits rather poorly into the adjacent 5-membered chelate rings of the tridentate ligand. This is especially evident from the trans Npz–Ni–Npz angles, which deviate significantly from 180° and measure 155.53(7) and 154.52(4)° for 7 and 8, respectively. These values match the mean Npz–Ni–Npz angle for all crystallographically characterized Ni(II) complexes of 2,6-bis(pyrazole)pyridine currently available in the CSD25 (Table 1). The same observation holds for the Npy–Ni–Npz bond angles. Inspection of the data in Table 1 reveals that there is surprisingly little variation in the bond angles subtended at the metal ion for this class of compounds, consistent with an essentially inflexible chelating ligand. The fact that the mean endo-Npy–Ni–Npz bond angle deviates significantly from 180° reflects the geometric constraints of the ligand framework which precludes attainment of the an “ideal” trans-Npy–Ni–Npz bond angle of 180° for a six-coordinate Ni(II) complex.
|
||||
|---|---|---|---|---|
| Compound | Ni–Npy | Ni–Npz | Npz–Ni–Npz | Npy–Ni–Npz |
| a Abbreviations: CSD, Cambridge Structural Database; Npy, pyridyl nitrogen atom; Npz, pyrazole nitrogen; s.u., standard uncertainty. | ||||
| KUFTUB | 2.023 | 2.132 | 153.3 | 76.8 |
| 2.112 | 76.5 | |||
| KUFVAJ | 2.036 | 2.113 | 153.5 | 76.7 |
| 2.104 | 76.8 | |||
| NASTAD | 2.026 | 2.109 | 152.9 | 76.7 |
| 2.099 | 76.7 | |||
| NEQZUE | 2.012 | 2.101 | 153.8 | 76.8 |
| 2.014 | 2.123 | 153.8 | 77.1 | |
| 2.119 | 76.9 | |||
| 2.094 | 77.1 | |||
| NERBER | 1.995 | 2.106 | 155.7 | 78.5 |
| 1.985 | 2.116 | 155.9 | 77.2 | |
| 2.122 | 77.7 | |||
| 2.137 | 78.2 | |||
| OFOTIN | 2.005 | 2.064 | 154.9 | 77.6 |
| 2.091 | 77.4 | |||
| 7·4H2O | 2.005 | 2.083 | 155.5 | 77.6 |
| 2.078 | 78.0 | |||
| 8·2H2O | 2.016 | 2.059 | 154.5 | 77.6 |
| 2.101 | 77.1 | |||
| mean | 2.012 | 2.103 | 154.4 | 77.2 |
| s.u. | 0.015 | 0.021 | 1.1 | 0.5 |
| min. | 1.985 | 2.059 | 152.9 | 76.5 |
| max. | 2.036 | 2.137 | 155.9 | 78.5 |
The strained coordination geometry for 7 and 8 has possibly significant structural consequences for the aqua ligands. In 7, the axial Ni–OW distances average 2.096(24) Å and are considerably longer than the in-plane coordination distance of 2.041(2) Å (Ni1–O1). The closer approach of the in-plane water molecule is evidently favoured by reduced steric encumbrance from the widened in-plane exo-Npy–Ni–Npz bond angle (205°). A similar disparity exists for the Ni–OW bond distances of 8 for the same reason. Since catalysis by these complexes (assuming that they might be partially or fully aquated in 2-propanol when aqueous KOH or NaOH are used as bases) would require the exchange of Ni-bound water molecules for ketone, alcohol, or alkoxide substrates, it is possible (assuming further that the Ni–O bond length is proportional to reactivity) to speculate that substitution of the sterically-accessible axial water molecules at the metal centres of 7 and 8 precedes substitution of the in-plane aqua ligand. In both 7 and 8, the Ni–Npz bonds are inequivalent, fall in the range 2.005–2.101 Å and average 2.080(17) Å. The central Ni–Npy bonds are somewhat shorter, averaging 2.011(8) Å for the two complexes. From the data compiled in Table 1, it is clear that the Ni–Npy bonds are normal for this class of compounds, matching the mean of 2.012(15) Å. Although the bonds between the metal ion and pyrazole nitrogen atoms in 7 and 8 are shorter than the mean [2.103(21) Å] for this class of chelates, they are equivalent within one standard deviation of the mean (1σ). Because of the rigidity of the 2,6-bis(pyrazole)pyridine Ni(II) chelate for all entries in Table 2, the relative invariance of the Ni–N bonds is to be expected in this class of compounds and, furthermore, appears to be insensitive to whether the pyrazole moieties are alkylated or not. Finally, it is noteworthy that the chloride salt 8·2H2O is structurally and crystallographically isomorphous with the bromide salt reported by Tastekin et al.,26 namely diaqua-bromo-(2,6-bis(3,5-dimethyl-pyrazolyl)pyridine-N,N′,N′′)-nickel(II) bromide.
| Catalyst | Conv. 48 h (%)b | 105 kobs (s−1) | R2 (fit) |
|---|---|---|---|
| a Conditions: acetophenone, 2.00 mmol; catalyst, 0.02 mmol (1.0 mol%); base, 0.40 M KOH in 2-propanol (5 mL); temperature, 82 °C.b Determined by 1H NMR spectroscopy at t = 48 h. | |||
| 1 | 95 | 3.4(5) | 0.983 |
| 2 | 85 | 1.6(3) | 0.980 |
| 3 | 96 | 3.5(3) | 0.988 |
| 4 | 94 | 2.2(5) | 0.951 |
| 5 | 70 | 4.3(2) | 0.999 |
| 6 | 94 | 3.7(5) | 0.986 |
| 7 | 96 | 0.88(31) | 0.964 |
| 8 | 99 | 2.1(5) | 0.952 |
| 9 | 99 | 2.9(5) | 0.970 |
Transfer hydrogenation (TH) of ketones
Complexes 1–9 were investigated as catalysts for the TH of acetophenone as a model substrate using KOH as the base and 2-propanol as the hydrogen donor. The conversions of acetophenone to the product (1-phenylethanol) were determined by 1H NMR spectroscopy by comparing the intensity of the methyl signal of acetophenone (s, δ 2.59 ppm) and methyl signal of 1-phenylethanol (d, δ 1.49 ppm) of the crude product.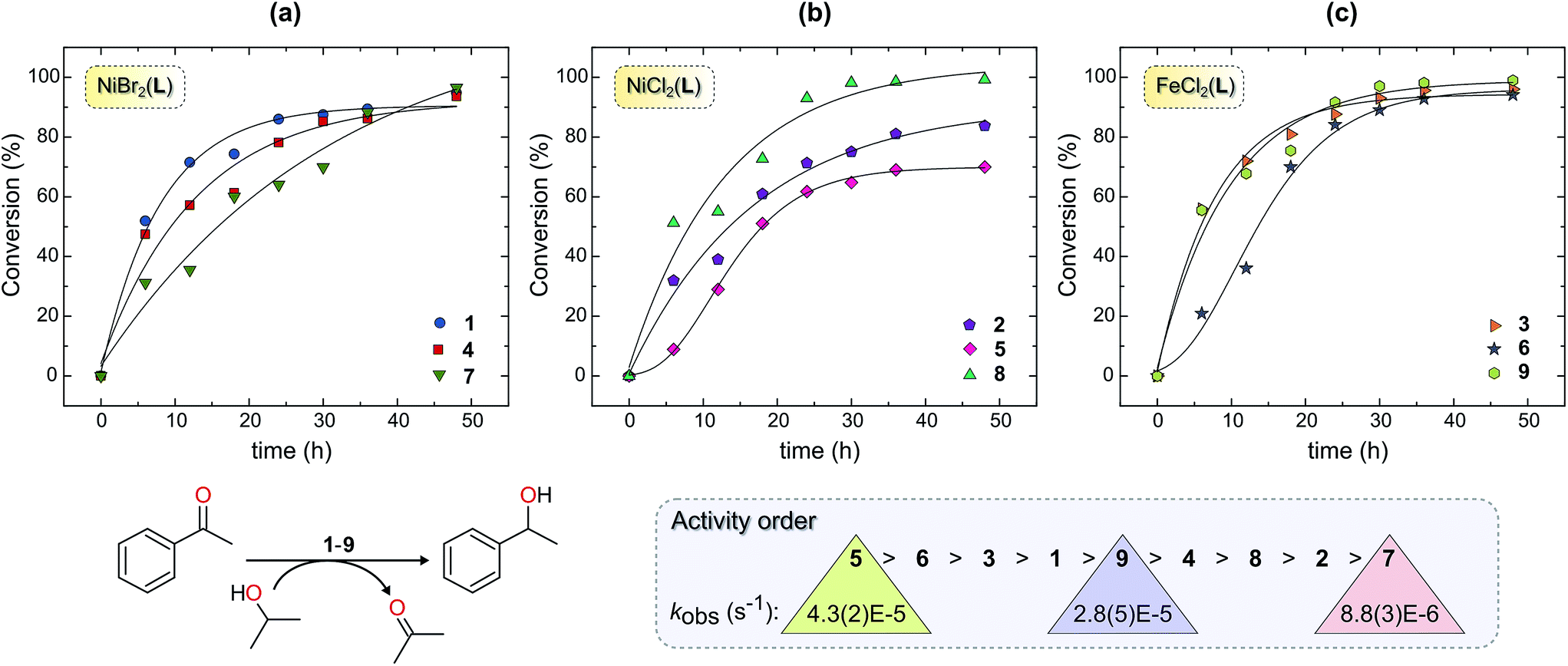 | ||
| Fig. 2 Time dependence of the catalytic transfer hydrogenation (TH) of acetophenone by complexes 1–9 in KOH/2-propanol at 82 °C. Graphs (a)–(c) show first-order exponential fits of the kinetics with the data grouped according to metal ion type, Ni(II) or Fe(II), and halide ion leaving groups (Br− or Cl−). The ligand L is either didentate L1 (compounds 1–3) or one of the tridentate ligands L2 (compounds 4–6) or L3 (compounds 7–9). The notably long induction phases for 5 and 6 necessitated use of the Gompertz model to fit the rate data. A steep initial rate coupled with incomplete conversion reflects an active catalyst that is susceptible to relatively rapid deactivation under the chosen reaction conditions. | ||
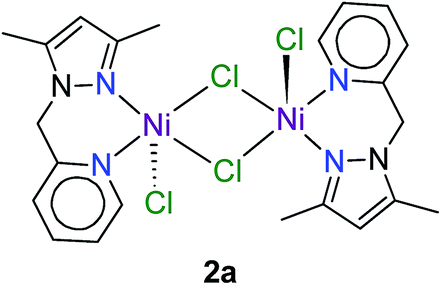
One explanation for the behaviour of 2 and 5 is ligand exchange and formation of catalytically inactive species. For 2, competing formation of a less-reactive dinuclear complex such as 2a during the reaction could, in principle, diminish turnover of the catalyst. Dinuclear halide-bridged species such as 2a are stable and, in the case of the bromide analogue, have been crystallo-graphically characterized.27 The marked deactivation of 5 evident in Fig. 2b is noteworthy, particularly in view of the high post-induction reactivity of the complex (cf. steep slope between 10 and 20 h). At present, we have no experimental data to account for this phenomenon, but note that both the formation of a dinuclear species and a 2:1 ligand:metal adduct (such as crystallographically characterized 4a in Fig. S2†) could drain (i.e., poison) the catalytically active species during the reaction, culminating in premature saturation of the kinetics.
Comparison of the iron and nickel complexes reveals that the Fe(II) derivatives afford catalytic species that encompass the more reactive half of the series delineated in Fig. 2. Despite 2 and 3 having the same ligand (L1), the activity of 3 (Fe2+ catalyst) was markedly higher than that of 2 (Ni2+ catalyst). This reactivity difference for the present series of complexes probably reflects the higher electropositivity and hardness of Fe(II) compared with Ni(II) and, consequently, the tendency of iron to form hydride intermediates28,29 from 2-propanol (as postulated for other TH catalysts)30,31 more readily than nickel. Despite their efficacy in the TH of acetophenone, the present Fe(II) chelates are relatively inactive compared with the Fe(II) carbonyl complexes reported by Morris and co-workers (which give 93% conversion to product in only 30 min).6,14 The present Ni(II) catalysts, on the other hand, exhibit activity profiles that more closely parallel the limited data available in the literature for homogeneous Ni(II) TH catalysts. For example, 1 has comparable activity (95% conversion; 48 h) to the Ni(II) complex of a PNO-donor chelate employed as an ATH catalyst reported by Dong et al. (98% conversion; 48 h).19
Further analysis of the structural and electronic factors that impact on the reactivity order of 1–9 in the TH of acetophenone is discussed within the context of our DFT simulations (vide infra). Noteworthy here is the fact that there appears to be no single dominant variable that succinctly accounts for the reactivity order of 1–9, attesting to an inherently complex mechanism. The exact catalytic species in 2-propanol for 1–9 have, furthermore, not been unequivocally established. For TH catalysts, which typically have induction periods, this is a notoriously difficult task, especially with paramagnetic complexes and intermediates. Complexes 1–9 are almost certainly precatalysts which have to undergo initial ligand exchange, e.g. displacement of metal-bound halide ions by the solvent or iPrO−, before M–H formation, substrate uptake, and thence hydrogen transfer. The X-ray structures of 7·4H2O and 8·2H2O are revealing in this regard because they highlight how two or three O-donor ligands may substitute the metal-bound halide ions of the initial metal complexes to give stable species. Halide-substituted species are evidently important intermediates in the present TH reactions, as discussed later.
| Entry | Substrate | Base | Conv. (%)b |
|---|---|---|---|
| a Conditions: ketone, 2.0 mmol; catalyst, 0.02 mmol (1.0 mol%); base, 0.4 M in 2-propanol (5 mL); time, 48 h; temperature, 82 °C.b Determined by 1H NMR spectroscopy.c 0.01 mmol catalyst (0.50 mol%).d 0.03 mmol catalyst (1.50 mol%). | |||
| 1 | 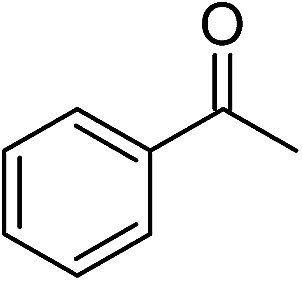 |
KOH | 95 |
| 2 | 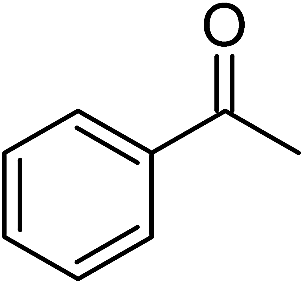 |
NaOH | 84 |
| 3 | 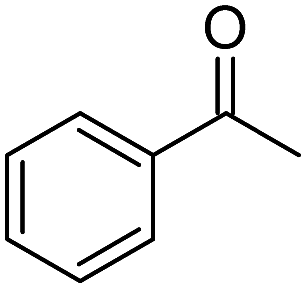 |
Na2CO3 | 55 |
| 4 | 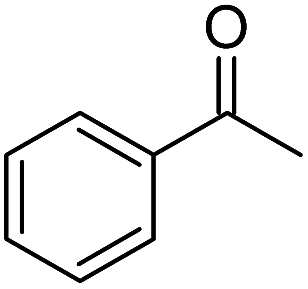 |
tBuOK | 97 |
| 5 | 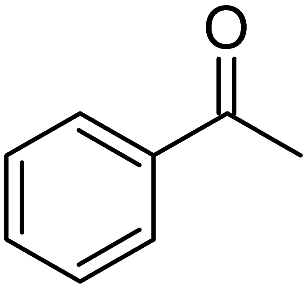 |
KOH | 60c |
| 6 | 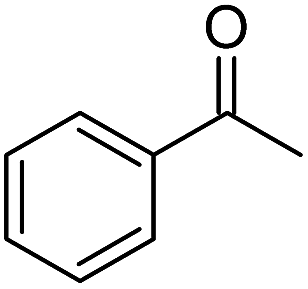 |
KOH | 59d |
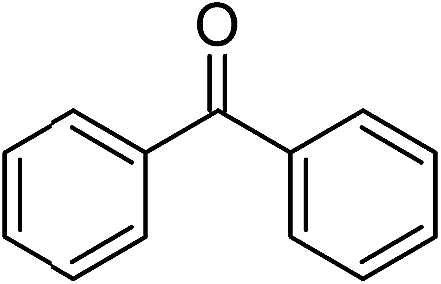
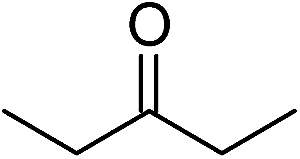
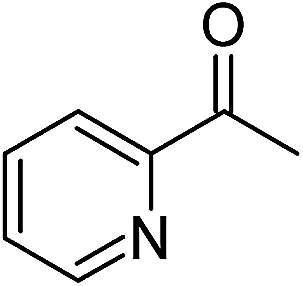
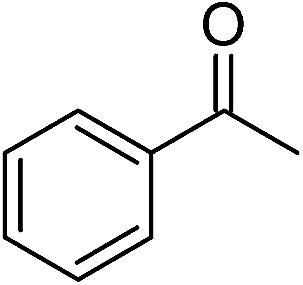
Significantly, we observed diminished catalytic activity for 2-methylcyclohexanone (77% conversion) and 3-pentanone (56% conversion) using 1 (or 9) as the catalyst. This suggests that 1 and 9 exhibit marked substrate specificity with aromatic ketones being favoured over aliphatic ketones. Even 2-acetylpyridine was efficiently reduced (96% conversion) by 9, indicating that potentially coordinating heterocyclic aromatic ketones may be equally efficiently reduced to the corresponding alcohol. Finally, benzophenone was the least favourable aromatic ketone substrate for 9 (89% conversion), consistent with increased steric hindrance to reduction engendered by the additional aryl ring appended to the carbonyl carbon.
DFT-simulated structures of 1–9
We have used DFT simulations at the HSEH1PBE/6-311g(d,p) level of theory in a 2-propanol solvent continuum to gain a conceptual understanding of the structures of the initial metal complexes in solution that are likely to be precatalysts in the transfer hydrogenation of acetophenone. A further goal was to attempt to understand the structural features of the complexes that might impact on the reaction kinetics.Table 5 summarizes selected bond distances and angles for 1–9; representative structures are given in Fig. 3, S7, and S8.† The Fe(II) chelates 3 and 6 have longer bond distances for the intrachelate interactions (M–Npy and M–Npz) than the Ni(II) complexes due to the larger ionic radius of high-spin Fe(II), 0.78 Å, relative to that of Ni(II), 0.69 Å.36 For complexes with didentate L1 (1–3), the M–X distances (for the same halide and metal ion) are about 5–7% shorter than for the complexes with the tridentate ligand L2. From Fig. 3, elongation of the M–X bonds for complexes of L2 reflects the increase in steric repulsion between the metal-bound halide ions and the methyl substituents of the coordinated pyrazole rings. As expected from the ionic radii of the halide ions within the two chelate categories (di- or tridentate), the M–Cl bonds are shorter than the corresponding M–Br bonds. Qualitatively, steric hindrance about the metal ion due to the chelating ligand's structure is amply highlighted by the structures shown in Fig. 3 and S8.† The metal ion is evidently less accessible to nucleophilic attack when the chelating ligand is tridentate L2. The impact of the chelating ligand's structure coupled with the size of the coordinated halide ion on the TH kinetics involving 1–9 is explored in considerable detail below in our attempts to delineate quantitative structure–activity relationships (QSARs) for the compounds.
| Cpd. | Symm. | M–X (Å) | M–Npy (Å) | M–Npz (Å) | Npy–M–Npz (°) | X–M–X (°) | Npy–M–X (°) | Q (M, e)c | ρS (M, e)d | GTM (complex) (%)e | ω (°)f |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Abbreviations: Symm.; symmetry of minimum energy structure; M = Ni or Fe; X = Cl or Br; Npy, pyridyl nitrogen; Npz pyrazolyl nitrogen.b Standard uncertainties are given in parentheses for bonds or angles where mean values are reported.c Partial charge (NBO) on the metal ion.d Unpaired spin density (Mulliken) localized on the metal ion. The total atomic spin density (all atoms) in each Ni(II) and Fe(II) complex summed to 2.00 and 4.00, respectively, consistent with the triplet and quintet spin states assigned in the simulation and the room temperature magnetic moments determined for 1–9 in this work.e The percentage of the metal ion's surface shielded by the ligand donor atoms in the theoretically-derived structure of the complex.f Equivalent cone angle, ECA, corresponding to the solid angle of the ligand. | |||||||||||
| 1 | C1 | 2.430(2) | 2.023 | 1.987 | 92.5 | 140.7 | 100.9(7) | 1.419 | 1.771 | 69.4 | 159.5 |
| 2 | C1 | 2.292(6) | 2.022 | 1.987 | 92.6 | 141.0 | 100(2) | 1.466 | 1.774 | 62.9 | 159.6 |
| 3 | C1 | 2.311(4) | 2.149 | 2.075 | 88.8 | 136.2 | 102.0(8) | 1.514 | 3.802 | 58.4 | 151.8 |
| 4 | C2 | 2.602(0) | 2.068 | 2.023(0) | 93.4(0) | 164.1 | 98.0(0) | 1.433 | 1.728 | 78.6 | 203.5 |
| 5 | C2 | 2.445(0) | 2.072 | 2.031(0) | 92.4(0) | 160.6 | 99.7(0) | 1.467 | 1.748 | 73.8 | 203.2 |
| 6 | C2 | 2.438(0) | 2.198 | 2.137(0) | 89.6(0) | 158.9 | 100.6(0) | 1.536 | 3.812 | 68.1 | 192.1 |
| 7 | C2 | 2.497 | 1.988 | 2.060 | 77.7 | 145.3 | 107.4(0) | 1.427 | 1.737 | 78.7 | 181.8 |
| 8 | C2 | 2.366 | 1.990 | 2.062 | 77.7 | 148.8 | 105.6(0) | 1.469 | 1.750 | 72.5 | 181.7 |
| 9 | C1 | 2.39(8) | 2.158 | 2.159(0) | 72.8 | 108.8 | 126(1) | 1.527 | 3.798 | 66.3 | 171.4 |
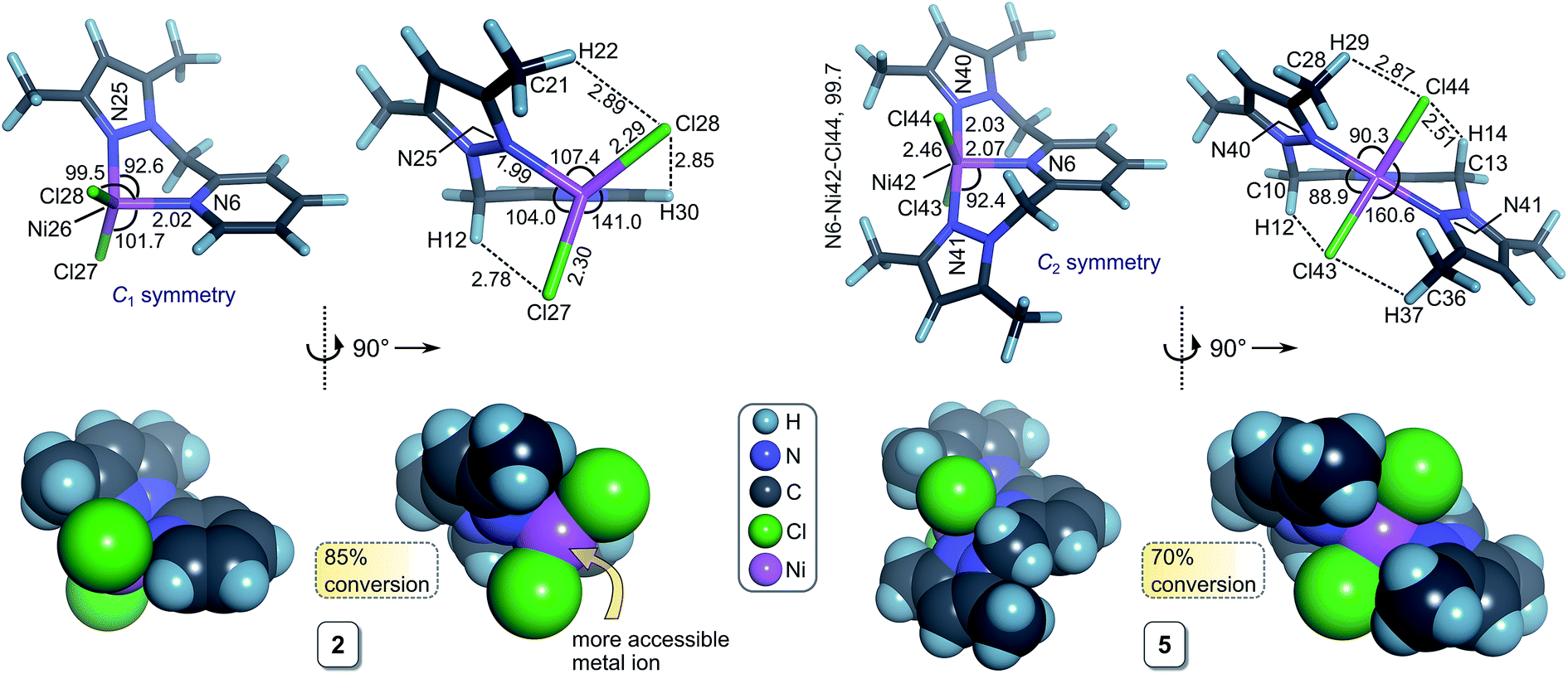 | ||
| Fig. 3 DFT-calculated geometries [HSEH1PBE/6-311g(d,p) level of theory; 2-propanol solvent continuum] of the active Ni(II) precatalysts 2 (C1 symmetry) and 5 (C2 symmetry) illustrating the different degrees of steric hindrance about the metal centre. The upper views show the optimized geometries as bond cylinder models with selected atom labels, bond distances (Å) and bond angles (°); the lower views depict the structures rendered with van der Waals radii for the atoms. The % conversion data refer to the TH reaction of acetophenone after 48 h. | ||
Since we have not been able to crystallize all of the precatalysts employed in this study, the question of the accuracy of the DFT simulations for structural simulations must be addressed. Unfortunately, few crystallographically characterized complexes with a single chelate ligand of the type L1 or L2 bound to either Ni(II) or Fe(II) exist in the literature for comparison with the DFT-calculated geometries of 1–9. A single, pertinent X-ray structure exists with L1 coordinated to Ni(II) for comparison, namely that of the related t-butyl-substituted complex dibromo-(2-((3,5-di-tert-butyl-1H-pyrazol-1-yl-N2)methyl)-pyridine-N)-nickel(II) (Fig. S9†).27 Comparison of the DFT-calculated structure of 1 with the foregoing experimental structure indicates that the coordination group bond distances are within 0.5% (M–N) to 2% (M–Br) of the experimental bond distances and that the ligand conformation is accurately reproduced. A second test of the DFT method and basis set is possible for compound 9; excellent agreement (bond distances, within 0.5% (M−N) to 4% (M–Cl); bond angles, <2% difference) between the DFT-calculated and experimental X-ray structure37 is evident from the least-squares fit wherein the rigid geometry of the chelating ligand of the X-ray structure is especially well-reproduced (Fig. S10†). The present simulations at the HSEH1PBE/6-311g(d,p) level of theory thus offer accurate structural parameters for the compounds investigated, an essential requirement for any attempt to delineate structure–activity relationships for the TH kinetics (vide infra).
Regarding the electronic structures of 1–9, the data in Table 5 confirm the high-spin states for the Ni(II) and Fe(II) compounds determined by magnetic susceptibility measure-ments on the powders. The unpaired spin density on the metal ion ranges from 1.728 to 1.774 e for the Ni(II) complexes and more narrowly from 3.798 to 3.812 e for the Fe(II) complexes. The partial cationic charge on the metal ion is relatively invariant, ranging from 1.419 to 1.469 e for the Ni(II) complexes and from 1.514 to 1.536 e for the Fe(II) complexes.
Of considerable interest for an analysis of the factors that might control the TH kinetics for reactions involving 1–9 is the degree of steric shielding of the metal ion. We used the program SOLID-G (2008) written by Guzei38 to calculate the ligand solid angles, equivalent cone angles, and overlap between the ligands for all final DFT-calculated structures of 1–9 in Table 5. The two most useful parameters calculated by the program are GTM (complex) (the percentage of the metal ion's surface screened or shielded by all of the donor atoms of the directly coordinated ligands in the theoretically-derived structure of the complex) and ω (the equivalent cone angle, ECA, for the chelating ligand determined from its specific coordinated geometry in the complex). The steric shielding parameter GTM (complex) indicates that the screening of the metal ion follows the expected order for the chelating ligands based on their structures and the degree to which they enwrap the metal ion (which may be separately quantified by ω): L1 < L3 < L2. This is paralleled by the expected order of GTM (complex) for the halide ions, Cl− < Br−, which reflects their ionic radii. Similarly, the Fe(II) complexes are less sterically shielded than their Ni(II) counterparts because of the 13% larger ionic radius of the Fe(II) ion,39 which accommodates the ligand donor atoms more spaciously over the surface of the metal ion sphere.
Kinetic QSARs based on DFT-calculated parameters
As noted above, acetophenone TH conversions (Table 2) ranged from ca. 70% to 99%; both the nature of the ligands and the metal ion were systematically varied to understand some of the factors that might contribute to the reactivity profiles of the compounds (precatalysts). The product yield data after 48 h for 2 (85%, didentate L1) and 5 (70%, tridentate L2) reflect the most pronounced ligand-dependence for the compounds studied. (Note that the rate constants in Table 2 and Fig. 2 measure mainly the initial catalyst activity and do not necessarily correlate with the reaction yields after 48 h, which are subject to yield-limiting factors such as catalyst deactivation, as discussed below.) From Fig. 3, it is clear that the metal centre of 5 is sterically more restricted than that of 2, principally due to crowding of the metal ion by the methyl groups (C28 and C36) of the two pyrazole moieties of tridentate L2. From Table 5, the metal ion steric shielding parameter, GTM (complex), quantitatively confirms the effect since only 62.9% of the metal ion's surface is shielded in the case of 2 compared with 73.8% in the case of 5. Increased steric crowding in the complex is evidently paralleled by commensurate elongation of the Ni–N and Ni–Cl bonds in 5 (by ca. 2.5% and 6.5%, respectively) relative to those for 2. The chloro ligands of 5 are, furthermore, more tightly held by intramolecular C–H⋯Cl hydrogen bonds involving, for example, H29 and H14. From Fig. 2b, it is clear that the kinetics for 2 follow a standard first order exponential growth curve (zero or negligible induction phase), while a long induction phase of ca. 2–4 h exists for 5. The kinetics for both 2 and 5 clearly exhibit incomplete conversion, in all likelihood due to catalyst deactivation, with the effect being most pronounced for 5. Interestingly, rate constants for 5 and 6 required fits of the data with the Gompertz model,40 which accounts for both induction and inhibition, the latter consistent with catalyst deactivation. The foregoing DFT calculations on the precatalysts (Fig. 3, Table 5) suggest that an explanation for the unique behaviour of both 5 and 6 (long induction phase, different kinetic model) compared with the remaining compounds involves both the increased steric restriction of the metal ion and the enhanced intramolecular H-bonding to the metal-bound halide ions (particularly in 5). These two factors evidently work in unison, slowing the rate of formation of an active catalyst species in which one or both halide ions are substituted by iPrOH (or iPrO−) on the pathway to attaining the transition state species. (Postulated transition states for TH catalysts have been reviewed by Morris and involve M–OCH(CH3)2 alkoxide species.28,41)Although one might anticipate that the initial reactivity of the precatalysts could be straightforwardly delineated from their structures, we found no linear correlation between the initial percentage conversion of acetophenone to product (t = 6 h) when the catalytic activity was plotted against either the DFT-calculated M–X bond distance or the equivalent cone angle, ECA (or ω), of the ligand (Fig. S11†). However, as shown by Fig. 4, the relationship between precatalyst structure and catalytic activity in the TH of acetophenone is more complicated, being simultaneously dependent on these two uncorrelated variables. The three-dimensional plot, a bivariate linear regression fit of the % conversion after 6 h as a function of the M–X bond distance and ω for the chelating ligand (where X = Cl or Br and M = Fe or Ni), shows that the initial activity of the complexes increases monotonically with increasing M–X distance, but decreases progressively with increasing ω. Since ω is a steric size parameter that effectively gauges the type of chelating ligand, the available data are cleanly differentiated into two groups (A and B). Group A complexes are chelated to the sterically less-encumbered ligand L1, which screens the metal ion to a smaller extent, giving commensurately less obtuse values of ω. Group B complexes, in contrast, are chelated by the more bulky, yet structurally-related tridentate ligand L2, which screens significantly more of the metal ion surface, as reflected by more obtuse ω values. From the DFT-calculated geometries of 1 and 4 (Fig. 4, Table 5), the more obtuse ω value for L2 is consistent with the tridentate nature of the ligand and the additional steric encumbrance of the metal ion caused by the methyl groups appended to the two pyrazole moieties of the ligand. Since the slope of the plot decreases with increasing ω (steric hindrance), the % conversion of the substrate is lower for more hindered complexes, consistent with expectation if steric effects influence the reactive intermediates.
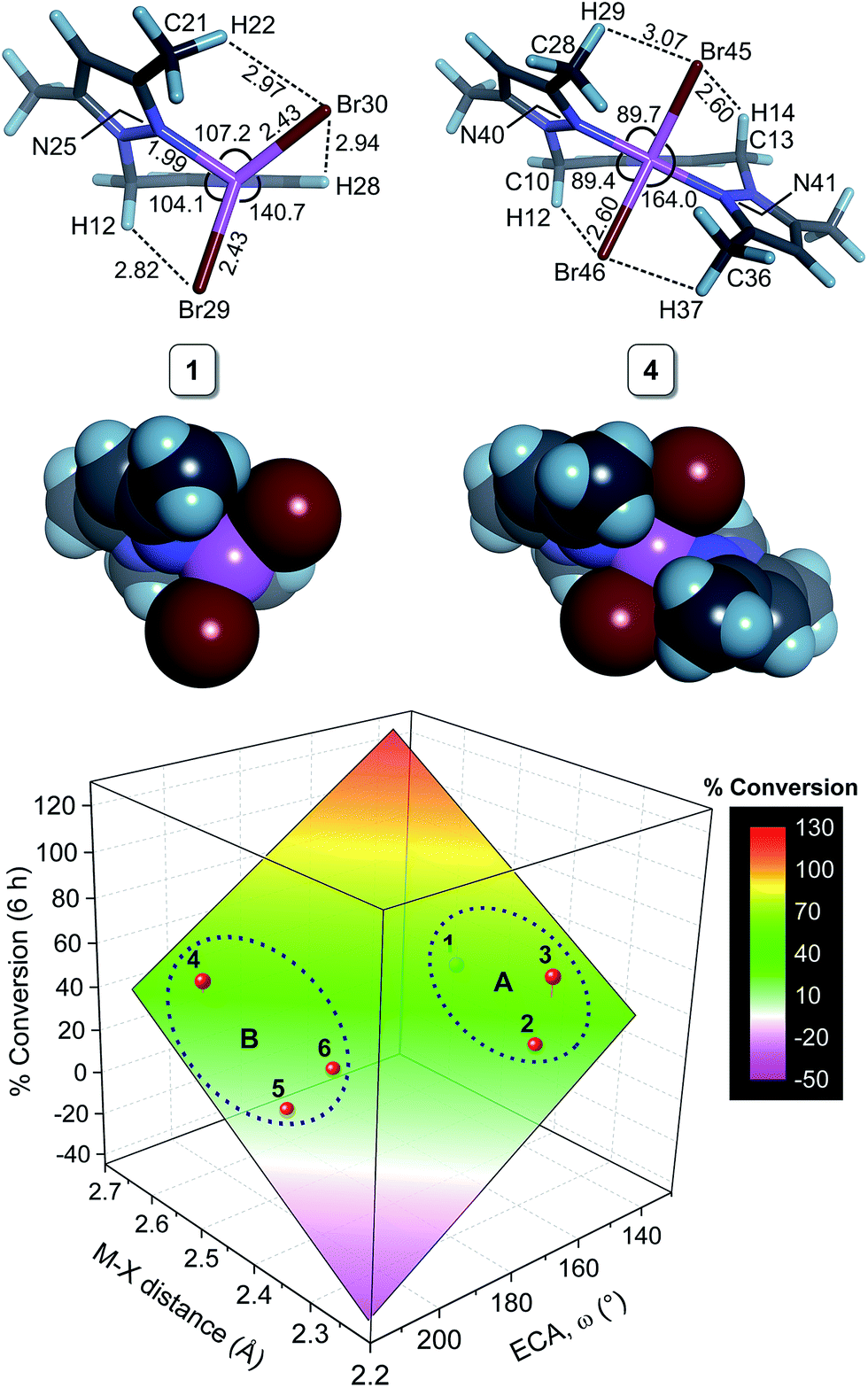 | ||
| Fig. 4 Top: Representative DFT-calculated structures of 1 and 4 in a 2-propanol solvent continuum (bond distances, Å; bond angles, °). Bottom: Analysis of the initial rate data for the TH of acetophenone by 1–6 as a function of the DFT-calculated M–X bond distance and the equivalent cone angle, ω, of the chelating ligand for the ligand systems L1 (group A) and L2 (group B). The plane represents a least-squares fit to the equation Z = z0 + ax + bx; % Conv. = −225(100) + 196(55)x – 1.2(3)y, where x = ECA and y = M–X distance (R2 = 0.800). The bivariate correlation reveals that the initial rate of transfer hydrogenation increases with increasing M–X bond distance and decreasing steric restriction of the metal centre by the chelating ligand. | ||
The significant 3D bivariate correlation of Fig. 4 did not, however, extend to complexes of L3, possibly because the latter tridentate ligand differs significantly from the former two in structure as it lacks the bridging methylene carbon and is an inherently more rigid, electronically-delocalized system. These (and possibly other) factors unique to L3 evidently impact on the initial rates of the TH reactions of 7–9 such that the QSAR delineated for 1–6 is not applicable to complexes of L3.
In Fig. 5 we have analysed the rate constant for TH of acetophenone, kobs, as a function of GTM (complex) and the M–Npy bond distance. The graphical illustration of GTM (complex) for 3, 9, and 4 shown in Fig. 5a highlights the effect that changing the size of the chelating ligand, halide ions, and metal ion has on the fraction of the metal ion's surface sterically shielded from substrate and solvent molecules in the reaction medium. Clearly, Fe(II) complexed to didentate L1 and two chloride ions (i.e., complex 3) will be the least sterically shielded, as discussed above (Table 5). The observed rate constant increases with decreasing GTM (complex) (i.e., reduced steric shielding of the metal ion) to some extent and, more significantly, with increasing M–Npy distance. For all complexes (excluding 1, 5, and 8) these dependencies underpin the 3D bivariate correlation of Fig. 5b. It is noteworthy that kobs shows effectively no independent linear correlation with GTM (complex), suggesting that steric shielding alone inadequately accounts for the TH rates.
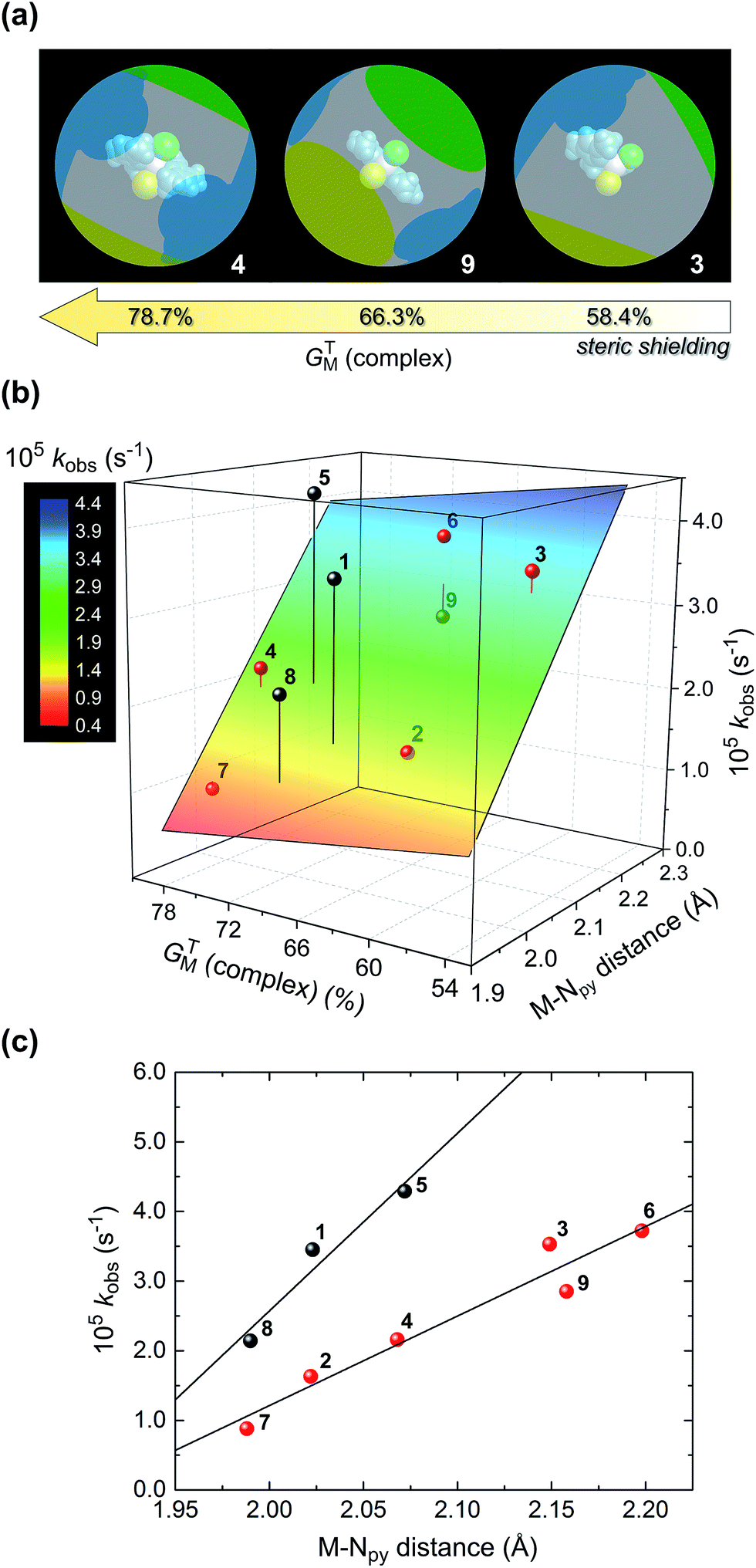 | ||
| Fig. 5 (a) Illustration of the degree of steric shielding (%) of the metal ion surface, GTM (complex), for 3, 9, and 4. The blue shadow represents screening by the chelating ligand; the green and yellow shadows reflect shielding by the upper and lower halide ions, respectively. (b) Graph of the rate constant for TH of acetophenone vs. GTM (complex) and the M–Npy bond distance. The plane is the least-squares fit of the data marked with red spheres; data with grey spheres are outliers. The equation of the plane, Z = z0 + ax + bx, is: % conv. = −2.1(5) × 10−4 + 1.2(2) × 10−4x – 1.9(1.9) × 10−7y, where x = M–Npy bond distance and y = GTM (complex) (R2 = 0.925). (c) Cross-section through the surface in part (b) showing the linear increase in rate constant with increasing M–Npy bond length. The linear equations are: kobs = −2.5(3) × 10−4 + 1.3(2) × 10−4(M–Npy), R2 = 0.924 (red spheres), and kobs = −5(1) × 10−4 + 2.6(6) × 10−4(M–Npy), R2 = 0.889 (grey spheres). | ||
However, kobs clearly correlates linearly with M–Npy distance. Furthermore, the complexes appear to be split into two independent groups (Fig. 5c). The reason for this is unclear, but the general trend is that longer M–Npy bonds lead to faster TH rates. This is readily understood in terms of the typical reactive species involved in TH reactions, namely metal hydrides coordinated cis to the ketone substrate in the case of inner-sphere reduction mechanisms.41–43 Based on the structures of the present compounds and the prevailing mechanisms in the literature, it is not unreasonable to suggest that elongation of the M–Npy bond promotes in-plane extrusion of the metal ion from the chelating ligand, thereby “priming” (exposing) the metal ion for formation of a reactive M–H species trans to the pyridyl nitrogen. Indeed, the calculated structures of the postulated hydride intermediates in the catalytic cycle (Fig. 6 and S12,† vide infra) confirm the existence of longer M–Npy bonds; complexes with intrinsically longer M–Npy bonds are thus likely to be more pre-organized for the formation of reactive hydrides. The analysis of the initial conversion efficacies above (Fig. 4) clearly reveals that the TH rates increase with increasing M–X distance. This is evidence that one or both of the halide ions dissociate from the metal ion to form the reactive species in this system. On the basis of our analysis of the precatalyst structures, initial rate data above, and the current literature on TH reaction mechanisms,41–44 it is possible to suggest candidate structures that might be relevant to the TH mechanisms of 1–9. For brevity, we have restricted this speculation to solution species derived from 8 and 9 because three key experimental insights exist (the X-ray structures of 8·2H2O and 937 and the substrate specificity data for 9 in Table 4) to guide our DFT simulations and ideas on reaction intermediates.
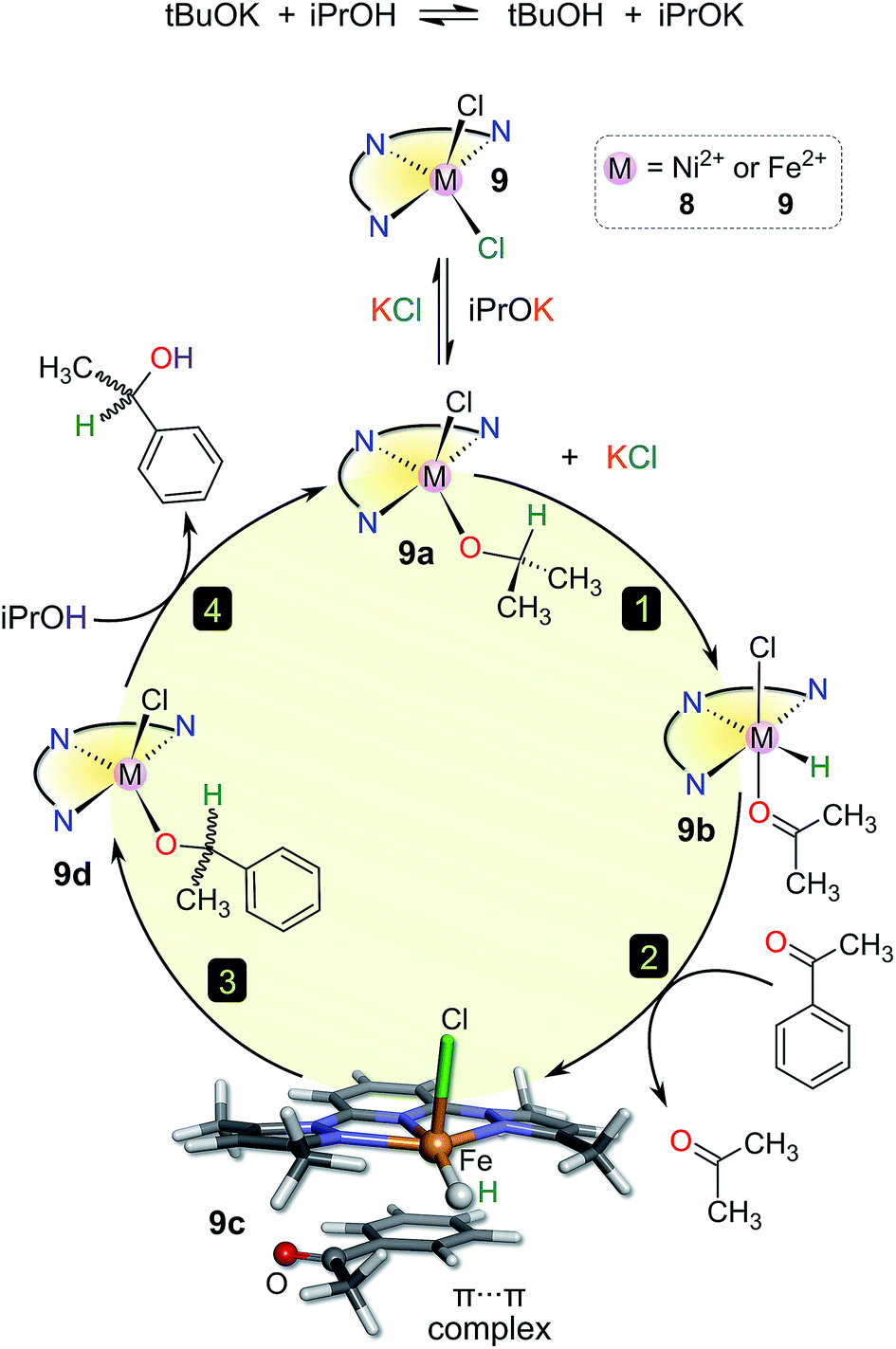 | ||
| Fig. 6 Postulated catalytic cycle for the TH of acetophenone by 8 or 9 in 2-propanol. Each DFT-calculated structure (see Fig. S12) in the cycle was a stable minimum at the HSEH1PBE/6-311G(d,p) level of theory in a 2-propanol solvent continuum. The cycle is illustrated using the high-spin Fe2+ complexes 9–9d as the species of interest. The structure of 9c was modelled by including an established empirical dispersion scheme in the functional. | ||
Possible mechanism
There are two commonly invoked transition state (TS) species in the TH of ketones when iPrOH is the reductant: (a) those derived from transient outer–sphere associations in which M–H and ligand X–H groups interact with the ketone carbonyl C- and O-atoms, respectively, typically in a pericyclic 6-membered ring (TS1), and (b) those derived from inner-sphere adducts, which generally involve coordination of the substrate ketone carbonyl group cis to a reactive M–H group (TS2).28,41 (The reactive M–H groups are formed by hydride transfer from metal-bound iPrO−, which subsequently dissociates as propanone.) Such states are depicted in Scheme 3 for transient species that could be derived from compounds 8 or 9 with a substrate such as acetophenone. (In Scheme 3, the tridentate ligand L3 is represented as the half-disk with pyridyl, py, and pyrazolyl, pz, nitrogen atoms marked.)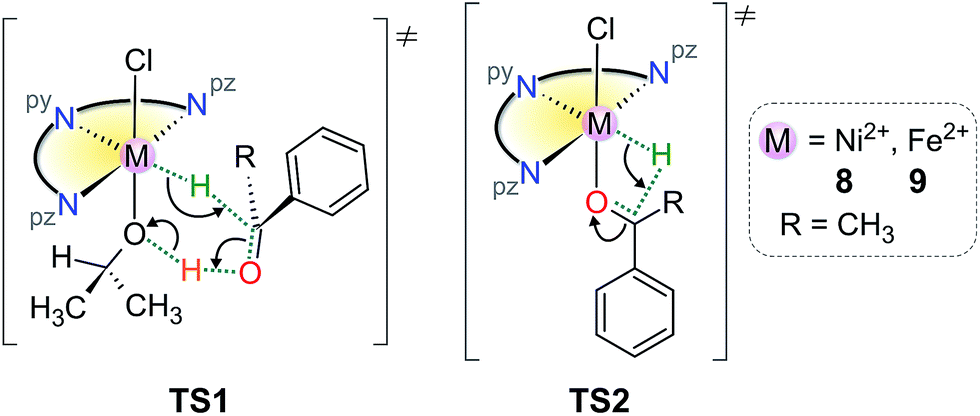 | ||
| Scheme 3 Typical transition states potentially of relevance to the TH mechanism of acetophenone by 8 or 9. | ||
In principle, the hydride ion could occupy any one of three meridional sites in these species, but substitution of chloride by iPrO− would be expected to place the O-bound iPrO− ion above or below the plane of L3 for steric efficiency and so the transfer of hydrogen from the iPrO− C–H donor to the metal ion most likely places the hydride ion in an equatorial position within the plane of L3. Such a site is only mildly sterically restricted by the flanking methyl groups from L3 and would readily accommodate a small anionic ligand. Formation of TS1 could then occur if a solvent molecule occupied the vacant site left by dissociating propanone; TS2 requires that acetophenone binds directly to the metal ion via its carbonyl oxygen to form an inner-sphere adduct.
Despite being reasonable postulates for the transition state structures involved in the TH of acetophenone with 8 or 9, neither transition state satisfactorily explains the large difference in the conversion efficiencies observed between aliphatic and aromatic ketone substrates (Table 4), mainly because no significant interaction between the ketone substituent groups and the catalyst exists in either TS1 or TS2. Indeed, our DFT simulations for 8 (Table S4†) suggest that there is no thermodynamic discrimination between aliphatic and aryl ketone substrates during the formation of the pre-transition state structure of TS2 (which is predicted to be endergonic at 298 K).
As noted earlier, irrespective of the ring substitution pattern, all ketone substrates with a single aromatic ring are practically completely converted (96–99%) to the corresponding alcohol product. An explanation for this key observation is that the pre-transition state structure for TH is unique for this system and likely involves formation of a stable π-adduct between the aromatic ligand of the catalyst and the ketone substrate. (Aromatic ketones would be expected to promote the formation of a π-adduct, favouring higher turnover of the reaction.)
A possible catalytic cycle for the system that reflects the experimental facts with 9 is depicted in Fig. 6 (ideal anhydrous conditions, tBuOK as base). DFT-calculated structures for 9–9d are given in detail in Fig. S12.† Briefly, the role of the added base (tBuOK) is to deprotonate iPrOH; the position of this equilibrium, and thus the effective concentration of iPrOK, is dependent on the strength of the base (Table 3). Precatalyst 9 reacts with iPrOK to form 9a by substitution of the coordinated halide ion, consistent with the dependence of the initial yield of product on the M–X bond distance (Fig. 4) and the observation that more weakly-bound halide ions favour higher initial rates. Compound 9a is the active catalyst; hydride migration from the methine CH group of the metal-bound iPrO− ion in step 1 affords the reactive metal hydride species 9b, as with many other TH catalysts.4,41 Transient coordination of the oxidized product (propanone) is possible (though not obligatory) given the inner-sphere nature of the hydride transfer step. Uptake of the aryl ketone substrate as a π-adduct in step 2 displaces propanone and forms the stable species 9c. The fact that aliphatic ketone substrates are poorly converted to product (Table 4) suggests that formation of a π-adduct (enhanced by the presence of an aryl ring) is important in the catalytic cycle of this system. The ketone carbonyl oxygen is positioned close to the reactive M–H group in 9c, facilitating nucleophilic hydride transfer to the carbonyl carbon in step 3. The addition product (an aryl alkoxide) binds to the metal ion via its O− donor (9d). In the final step, isopropyl alcohol protonates the metal-bound aryl alkoxide to release the reduction product (racemic 1-phenylethanol), regenerating the catalyst, 9a. One mole of iPrOH is therefore consumed for each mole of ketone reduced.
Fig. 7 highlights the structure of π-adduct 9c in more detail due to its relevance to the experimental substrate specificity of 9 (Table 4). Significantly, 9c is characterized by an offset interaction of the pyridine ring of the chelate with the aryl ring of the ketone substrate (i.e., a 1.91 Å lateral shift). The ring centroid separation, Cg1⋯Cg2, and perpendicular displacement measure 3.50 Å and 2.93 Å, respectively, and the associated slip angle is 33.1°. Note that the structure was modelled using the established empirical dispersion scheme from the APFD functional45 to augment the HSEH1PBE functional46 used here. Based on the experimental metrics of π–π stacking in metal–pyridyl complexes reported in Janiak's seminal review of the subject,47 wherein offset parallel stacking (laterally-shifted aromatic rings) is emblematic, centroid⋯centroid separations typically range from 3.4 to 4.2 Å (tight interactions), and slip angles average 27° for over 7600 structures, the simulated structure of π-adduct 9c is consistent with X-ray data for several thousand π-stacked metal–pyridine ring systems.
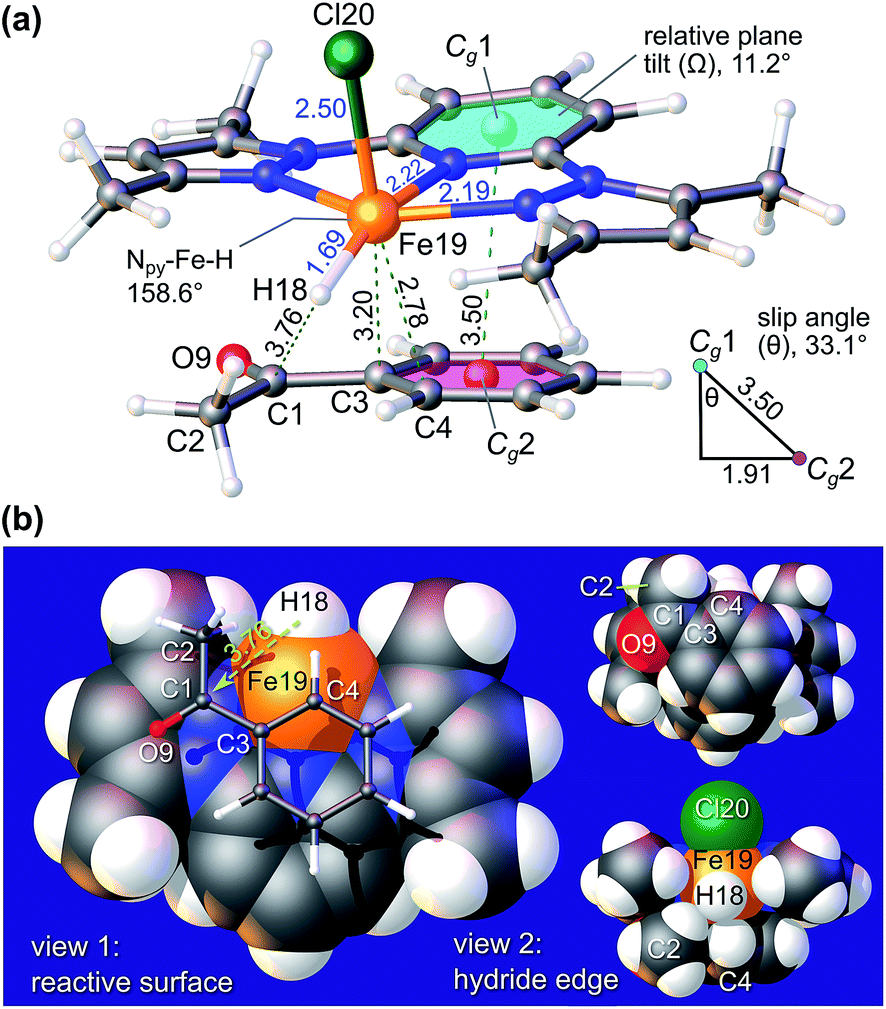 | ||
| Fig. 7 (a) DFT-calculated structure (dispersion effects included) of 9c, the pre-transition state π-adduct formed between the postulated hydride intermediate of 9 and acetophenone during the TH catalytic cycle. The geometric relationship between the pyridine ring plane of the catalyst and the aryl ring of the substrate is shown. (b) Selected views of the π-adduct with atoms rendered at their van der Waals radii. In both parts, key bond distances and contacts are given in Å and selected atoms are labelled. | ||
Because the two aromatic rings exhibit a tilt angle Ω of 11.2°, the reactive hydride ion (H18) is canted towards the carbonyl carbon (C1) of the substrate such that the H18⋯C1 distance is only 3.76 Å in the pre-transition state structure. A second noteworthy feature of 9c is the short interaction distance (2.78 Å) between Fe19 and C4 of the benzophenone phenyl ring. This distance is significantly less than the sum of the van der Waals radii of Fe (2.05 Å) and C (1.70 Å),48 and, based upon the NBO-calculated49 partial charges of 9c (Fig. S13†), may be regarded as a significant electrostatic attraction between Fe (δ, +1.446e) and C4 (δ, −0.264e). Evidently, π-adduct 9c is stabilized by well-defined attractive electrostatic interactions, dispersion forces between the aromatic rings, and a favourable frontier molecular orbital (FMO) interaction. The latter seemingly involves overlap of the highest singly-occupied MO (HSOMO, with predominantly dx2−y2 and σ-symmetry Fe–H bond character) and the LUMO (π*) of acetophenone (Fig. S14†). The interaction stabilizes the HSOMO of 9c by 0.155 eV (14.9 kJ mol−1) relative to the energy of this orbital in the non-interacting hydride, FeCl(H)(L3), and provides a familiar basis for understanding the association of FeCl(H)(L3) with acetophenone. From Fig. 7b, simple lateral translation (surface slippage) of the ketone carbonyl carbon atom (C1) towards the metal-bound hydride ion (H18) would ostensibly culminate in a TS from which SN2-type nucleophilic attack of C1 by the hydride ion with concerted formation of an O–Fe bond would afford the corresponding iron-bound aryl alkoxide 9d. Protonation of 9d by iPrOH in the last step of the catalytic cycle gives the aryl alcohol reduction product and regenerates the catalyst (Fig. 6).
The exact nature of the TS for step 3 in the catalytic cycle unfortunately remains elusive; numerous attempts to locate the TS using standard computational methods were unsuccessful. Part of the problem is that the initial (9c) and final (9d) structures linked by the TS on the reaction coordinate for step 3 have to be calculated with different model chemistries. Thus, while a functional with an empirical dispersion correction is mandatory for accurate calculation of π-adduct 9c, the same functional is in fact deleterious to structural simulation of 9d. This limitation aside, it is noteworthy that the aryl ketone may form the π-adduct by stacking with either face of the aryl ring in contact with the accessible face of the catalyst (which naturally orients the carbonyl group to the left or right of the M–H group). Consequently, the resulting two mirror image forms of 9c will give equivalent yields of the two enantiomers of 1-phenylethanol if the TS for step 3 retains some of the π-stacked character of 9c. An intermediate such as 9c in the catalytic cycle clearly accounts for the racemic product observed in the reaction.
Finally, thermochemical analysis (HSEH1PBE/6-311G(d,p) level of theory, 2-propanol solvent continuum) of the catalytic cycle for 9 (Fig. 6) using the geometry-optimized structures of all species in the reaction at 298.15 K affords several significant insights (Fig. 8). First, the induction step for 9, which involves substitution of iron-bound chloride by iPrO− is endergonic (+85.3 kJ mol−1); this reflects the experimental fact that the reaction only proceeds at elevated temperatures (refluxing 2-propanol). Second, the intramolecular hydride transfer step in which the π-adduct 9c converts to the aryl alkoxide 9d is highly exothermic (step 3, −189.2 kJ mol−1). Step 3 in the cycle clearly acts as the thermodynamic driving force needed to pull the system through the preceding endergonic steps. Third, the final step in which the iron-bound aryl alkoxide is protonated by iPrOH in solution to regenerate the active catalyst 9a and the product (1-phenylethanol) is only mildly endergonic (+7.0 kJ mol−1). The net Gibbs energy for the full reaction including the induction (activation) step amounts to 91.4 kJ mol−1; the in-cycle thermodynamics (reaction coordinate states 2 through 6) are commensurately more favourable with a net Gibbs energy of +6.0 kJ mol−1, but nevertheless still endergonic.
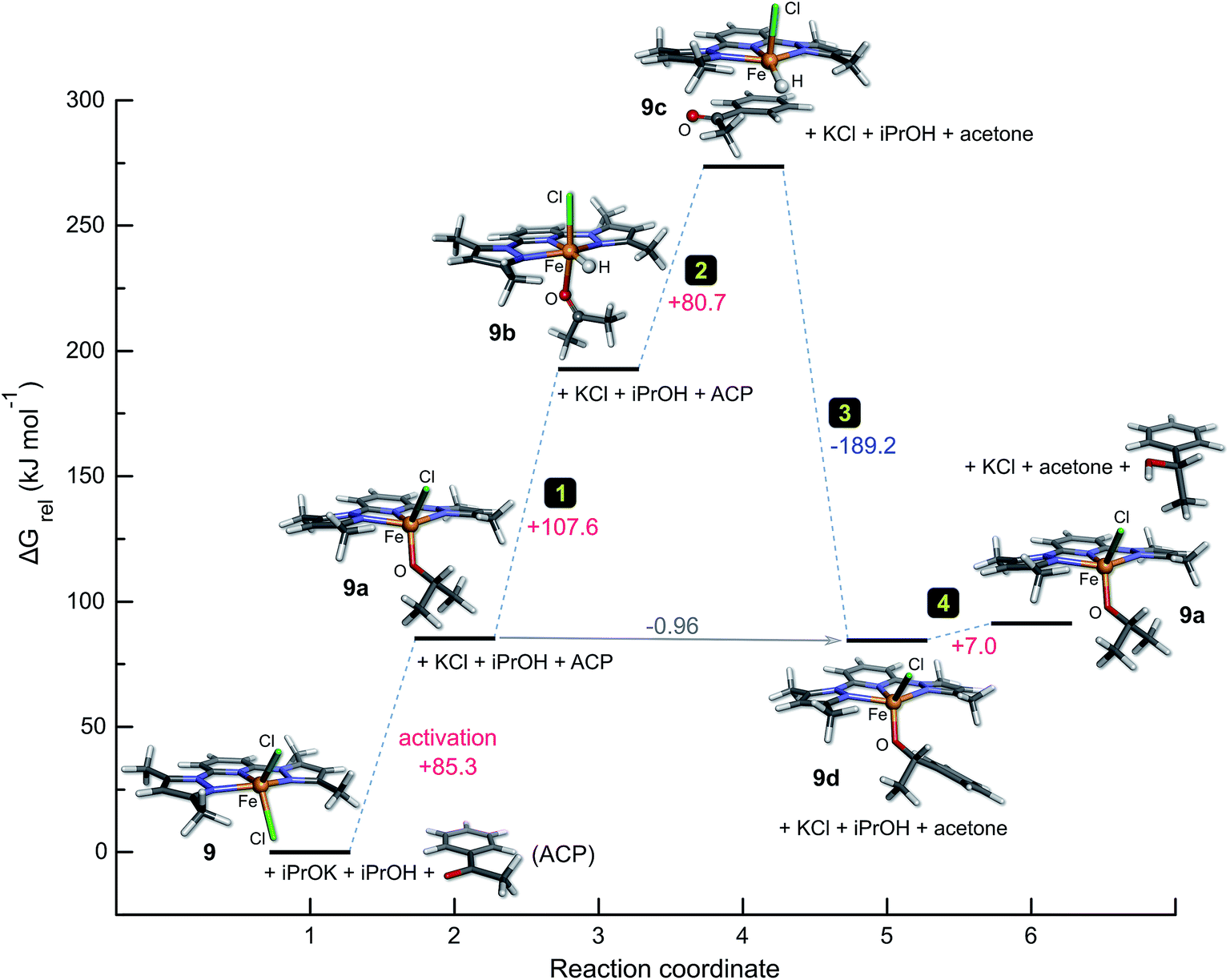 | ||
| Fig. 8 Graph of the Gibbs energy as a function of the reaction coordinate for the TH of acetophenone by 9 in 2-propanol. Thermochemical analysis including zero point vibrational energy corrections to the total energy for all species was performed at 298.15 K (P = 1.0 atm) with geometry-optimized minima at the HSEH1PBE/6-311G(d,p) level of theory. Steps corresponding to the full catalytic cycle in Fig. 6 are indicated; data used to construct the graph are given in Tables S5 and S6 (ESI†). | ||
Overall, the calculated Gibbs energies for the postulated catalytic cycle (with 9 as the case study) account adequately for the induction phases observed to a lesser or greater extent in the experimental reaction kinetics of several of the complexes investigated here (Fig. 2) as well as the fact that TH reactions of acetophenone with 1–9 require relatively long reaction times at 82 °C in 2-propanol for optimal conversions to product. Lastly, although mechanistically distinct, the orders of magnitude of the Gibbs energy changes computed here for the catalytic cycle of 9 are broadly in accord with the DFT-calculated Gibbs energy changes reported by Landwehr et al.50 for bifunctional rhenium cyclopentadienyl-type TH catalysts (despite the different model chemistry, catalysts, and conditions used in the simulations by these authors).
Conclusions
Iron(II) and nickel(II) complexes of bidentate (pyrazolylmethyl)pyridine (L1) and tridentate bis(pyrazolyl-methyl)-pyridine (L2) or bis(pyrazole)pyridine (L3) type ligands have been prepared, characterized, and tested as transfer hydrogenation (TH) catalysts for the reduction of ketones to secondary alcohols. The 1:1 ligand:metal complexes were moderately active catalysts for the TH of ketones. In most (but not all) cases, the Fe(II) complexes were more active than their Ni(II) analogues possibly because Fe(II) is harder than Ni(II), thereby favouring the formation of alkoxide and hydride intermediates. The catalytic performance was dependent, in a complex way, on the ligand structure, the metal-bound halide ions, and the nature of the ketone substrate (aryl ketones were preferable substrates to aliphatic ketones).
A combination of solid angle calculations (measuring steric shielding of a metal ion in a ligand field) and DFT simulations were used to delineate some of the key structural and electronic parameters of 1–9 that have an impact on the initial conversion rates and overall substrate specificities of the catalysts. Our analysis reveals that the initial conversion rates increase with increasing metal–halide bond distance and decreasing steric shielding of the metal ion for 1–6 (ligands L1 and L2). Furthermore, the observed rate constants, kobs, for TH of acetophenone increased with decreasing steric shielding of the metal ion and increasing M–Npyridine bond distance. These observations, in conjunction with the strong preference of catalysts 1 and 9 for aryl ketone substrates, suggest that a simple 4-step catalytic cycle (after endergonic induction) adequately accounts for the data. A key intermediate in this cycle is a stable π-adduct (9c) formed between the chelating pyridyl ligand of the catalyst and the aromatic ketone substrate; 9c accounts for both the substrate specificity of 9 and the racemic alcohol product, (R,S)-1-phenylethanol, generated in the reduction of acetophenone.
Experimental methods
General methods
All reagents and solvents were obtained from Sigma-Aldrich. The starting materials NiCl2, NiBr2, FeCl2·4H2O, isopropanol, absolute ethanol and deuterated solvents were used as received without further purification. Literature procedures were used to synthesize the free ligands 2-pyrazolyl(methyl)pyridine (L1),27 2-6-bis-(pyrazolyl-methyl)-pyridine (L2),27 and 2,6-bis(3,5-dimethyl-N-pyrazolyl)-pyridine (L3).51 Dichloromethane was dried over P2O5 and distilled prior to use. NMR spectra were recorded on a Bruker Avance 400 MHz (1H) [100 MHz (13C)] NMR spectrometer. All chemical shifts, δ, are reported in (ppm). Elemental analyses were performed on Thermo Scientific™ Flash 2000 Analyzer and mass spectra were recorded on a Waters® Micromass LCT Premier™ Mass Spectrometer. Magnetic moment measurements were performed with a Sherwood Scientific MK 1 Magnetic Susceptibility Balance.Synthesis of iron(II) and nickel(II) complexes
The synthetic procedures used to synthesize metal chelates of L1–L3 were essentially straightforward, as described below.:C, 32.56; H, 3.23; N, 10.36. Found C, 32.18; H, 4.57; N, 10.06. μeff = 2.89 BM.:C, 36.10; H, 3.87; N, 11.70. Found C, 35.86; H, 5.56; N, 11.99. μeff = 3.05 BM.Efforts to recrystallize 4 from powder re-dissolved in CH2Cl2 and layered with hexane afforded the 2:1 ligand:metal adduct [Ni(L2)2]Br2, presumably as a result of ligand exchange, eqn (1):
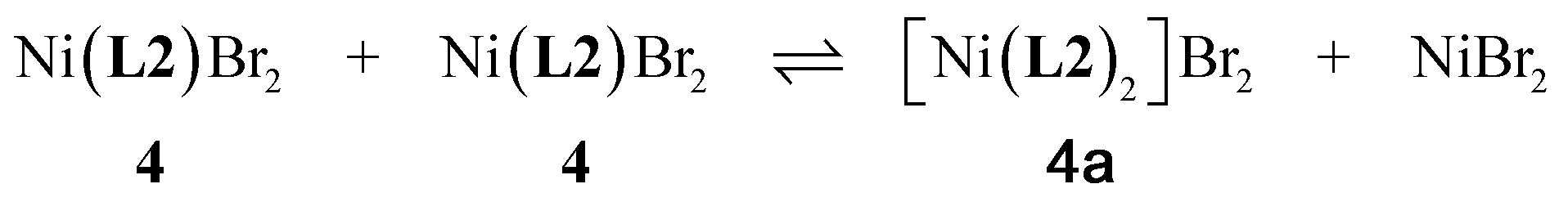 | (1) |
The presence of excess free ligand (L2) in the crystallization solution could, if present, also result in the formation of 4a. The molecular structure of 4a is given in Fig. S2–S4 (ESI†).
Despite attempts to recrystallize 6 from powder re-dissolved in CH2Cl2 and layered with hexane, only the 2:1 ligand:metal adduct was isolated in single crystal form suitable for X-ray diffraction analysis. Formation of the salt [FeII(L2)2][FeIIICl4]2, 6a, during crystallization evidently reflects both ligand exchange and oxidation of some of the Fe(II) present in the system to Fe(III), culminating in the formation of the tetrahedral [FeIIICl4]− counter-ions. The molecular structure of 6a is given in Fig. S5 (ESI†).
X-ray crystallography
Single crystal X-ray diffraction studies for complexes 4a, 6a, 7, and 8 were carried out with a Bruker Apex II Duo equipped with an Oxford Instruments Cryojet operating at 100(2) K and an Incoatec IμS Mo micro source operating at 30 W power. The data were collected with Mo Kα radiation (λ = 0.71073 Å) at a crystal-to-detector distance of 50 mm. The following conditions were used for the data collection: omega and phi scans with exposures taken at 30 W X-ray power and 0.50° frame widths using APEX2.52 The data were reduced using outlier rejection, scan speed scaling, as well as standard Lorentz and polarization correction factors. A semi-empirical absorption correction was applied to the data in each case (SADABS53). Structures were solved with either SHELXS-97 (ref. 54) (direct methods) or charge-flipping (SUPERFLIP55) and refined with SHELXL-97 (ref. 54) within Olex2.56 All non-hydrogen atoms were located in the difference density map and refined anisotropically before including hydrogen atoms as idealized contributors in the least squares process. Their positions were calculated using a standard riding model with C–Haromatic distances of 0.93 Å (Uiso = 1.2Ueq), C–Hmethylene distances of 0.97 Å (Uiso = 1.2Ueq), and C–Hmethyl distances of 0.96 Å (Uiso = 1.5Ueq); methyl group orientations were fit to the experimental electron density distribution about the methyl carbon atom.The crystal of 6a was not twinned, despite possible twinning suggested by analysis of the structure factors. However, the two [FeCl4]− counter-ions were each disordered over two sites and were fit to a two-site disorder model in which the site occupancies were allowed to freely refine. For the counter-ion containing Fe2, the major component of the disordered anion had a site occupancy factor of 0.83(2); that for the anion containing Fe3 was 0.850(18).
086 reflections measured (4.246° ≤ 2Θ ≤ 75.582°), 16101 unique (Rint = 0.0197, Rσ = 0.0260) which were used in all calculations. The final R1 was 0.0304 (I > 2σ(I)) and wR2 was 0.0683 (all data).![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 13.7303(12) Å, b = 14.9885(13) Å, c = 15.6987(13) Å, α = 89.620(4)°, β = 64.621(4)°, γ = 63.481(4)°, V = 2542.7(4) Å3, Z = 2, T = 100.0 K, μ(MoKα) = 1.514 mm−1, Dcalc = 1.583 g cm−3, 36960 reflections measured (2.95° ≤ 2Θ ≤ 52.468°), 9820 unique (Rint = 0.0284, Rσ = 0.0250) which were used in all calculations. The final R1 was 0.0966 (I > 2σ(I)) and wR2 was 0.2466 (all data).399 reflections measured (4.448 ≤ 2Θ ≤ 60.958), 4201 unique (Rint = 0.0210, Rσ = 0.0287) which were used in all calculations. The final R1 was 0.0150 (I > 2σ(I)) and wR2 was 0.0368 (all data).261 reflections measured (5.82 ≤ 2Θ ≤ 61.072), 5429 unique (Rint = 0.0197, Rsigma = 0.0163) which were used in all calculations. The final R1 was 0.0212 (I > 2σ(I)) and wR2 was 0.0556 (all data).
(no. 2), a = 13.7303(12) Å, b = 14.9885(13) Å, c = 15.6987(13) Å, α = 89.620(4)°, β = 64.621(4)°, γ = 63.481(4)°, V = 2542.7(4) Å3, Z = 2, T = 100.0 K, μ(MoKα) = 1.514 mm−1, Dcalc = 1.583 g cm−3, 36960 reflections measured (2.95° ≤ 2Θ ≤ 52.468°), 9820 unique (Rint = 0.0284, Rσ = 0.0250) which were used in all calculations. The final R1 was 0.0966 (I > 2σ(I)) and wR2 was 0.2466 (all data).399 reflections measured (4.448 ≤ 2Θ ≤ 60.958), 4201 unique (Rint = 0.0210, Rσ = 0.0287) which were used in all calculations. The final R1 was 0.0150 (I > 2σ(I)) and wR2 was 0.0368 (all data).261 reflections measured (5.82 ≤ 2Θ ≤ 61.072), 5429 unique (Rint = 0.0197, Rsigma = 0.0163) which were used in all calculations. The final R1 was 0.0212 (I > 2σ(I)) and wR2 was 0.0556 (all data).DFT simulations and structural analysis
Unless otherwise noted, DFT simulations were effected with Gaussian 09 Revision C.01 (WIN64)57 at the HSEH1PBE/6-311G(d,p) level of theory.46,58 A spin-unrestricted wave function was employed for all paramagnetic species (UHSEH1PBE model). Simulations were carried out both in vacuo and in a 2-propanol solvent continuum; the latter calculations employed the SCRF-PCM method.59 In all cases, frequency calculations were used to verify the nature of stationary states located on the potential energy surface; structures located and discussed in this work were true minima (as indicated by the absence of negative frequency eigen values). Population analysis of the final wave function for each geometry-optimized structure was effected with NBO 3.0 (ref. 49) running in Gaussian 09. Pre-transition state structures and all species involved in the catalytic cycles of 8 and 9 were calculated with Gaussian 09 Revision D.01 (WIN64) initially at the HSEH1PBE/SDD60 level of theory in 2-propanol (PCM) and then at the HSEH1PBE/6-311G(d,p) level of theory for publication. (The two methods gave similar results, with the exception of structure 9c, which was calculated to be more tightly interacting with the larger basis set.) The structure of the π-adduct 9c (Fig. 7) was calculated using the Petersson–Frisch empirical dispersion model from the APFD functional45 to augment the HSEH1PBE functional.Ligand steric effects in the DFT-calculated structures were quantified with Guzei's freely available SOLID-G program.38 In this program, the equivalent cone angle, ECA or ω, corresponds to the solid angle of the ligand. (Note that ω for the ligand is not the same as Tolman's cone angle for phosphine ligands.61)
Transfer hydrogenation reactions
Catalytic transfer hydrogenations of ketones were performed in two-necked round bottom flasks under nitrogen. In a typical experiment, 1–9 (0.02 mmol, 1%), 0.4 M KOH in 2-propanol (5 mL) and acetophenone (0.23 mL, 2.0 mmol) were added and refluxed at 82 °C under a static N2 atmosphere. Samples were taken at regular intervals and the course of the reaction was monitored by 1H NMR spectroscopy. The percentage conversions were calculated using the 1H NMR spectra by comparing the intensities of the methyl signal of acetophenone (s, δ 2.59 ppm) and the methyl signal of (R,S)-1-phenylethanol (d, δ 1.49 ppm) in the crude products. The solvent was removed under vacuum to obtain a brown product, which was isolated, recrystallized and characterized by 1H NMR spectroscopy.Kinetic data were analysed with 64-bit OriginPro 9.1.62 A standard nonlinear first order monomolecular exponential growth model, y = a(1 − e(−k(x−xc)),40 where a = amplitude, x = time, xc = centre, and k = rate, was used to fit the kinetic data for 1–4 and 7–9. Because of the significant induction phases evident for 5 and 6, the kinetic data for these two systems were analysed using the Gompertz model, y = ae−exp(−k(x−xc), where the parameters are as described above.40 This model is often applied to tumour or microbial cell growth as a function of time because it includes the effects of growth inhibition.63–65 The Gompertz model is thus ideal for empirical analysis of monomolecular product reaction kinetics wherein the trajectory describing growth in the reaction product (reduced ketone in this case) exhibits both induction and inhibition (i.e., catalyst deactivation).
Acknowledgements
The authors thank University of KwaZulu-Natal and the National Research Foundation of South Africa (NRF) for financial support. This work is based in part (O. Q. M.) on research supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation of South Africa (Grant No. 64799).Notes and references
- A. Zanotti-Gerosa, W. Hems, M. Groarke and F. Hancock, Platinum Met. Rev., 2005, 49, 158–165 CrossRef CAS
.
- R. M. Bullock, Angew. Chem., Int. Ed., 2007, 46, 7360–7363 CrossRef CAS PubMed
- D. Pandiarajan and R. Ramesh, J. Organomet. Chem., 2013, 723, 26–35 CrossRef CAS
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed
- L. Delaude, A. Demonceau and A. F. Noels, Curr. Org. Chem., 2006, 10, 203–215 CAS
- C. Sui-Seng, F. Freutel, A. J. Lough and R. H. Morris, Angew. Chem., 2008, 120, 954–957 CrossRef
- F. Zeng and Z. Yu, Organometallics, 2009, 28, 1855–1862 CrossRef CAS
- N. J. Beach and G. J. Spivak, Inorg. Chim. Acta, 2003, 343, 244–252 CrossRef CAS
- H. Deng, Z. Yu, J. Dong and S. Wu, Organometallics, 2005, 24, 4110–4112 CrossRef CAS
- R. M. Bullock, Chem.–Eur. J., 2004, 10, 2366–2374 CrossRef CAS PubMed
- C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816–5817 CrossRef CAS PubMed
- G. P. Boldrini, D. Savoia, E. Tagliavini, C. Trombini and A. Umani-Ronchi, J. Org. Chem., 1985, 50, 3082–3086 CrossRef CAS
- T. Hashimoto, S. Urban, R. Hoshino, Y. Ohki, K. Tatsumi and F. Glorius, Organometallics, 2012, 31, 4474–4479 CrossRef CAS
- N. Meyer, A. J. Lough and R. H. Morris, Chem.–Eur. J., 2009, 15, 5605–5610 CrossRef CAS PubMed
- S. Kuhl, R. Schneider and Y. Fort, Organometallics, 2003, 22, 4184–4186 CrossRef CAS
- F. Alonso, P. Riente and M. Yus, Tetrahedron Lett., 2008, 49, 1939–1942 CrossRef CAS
- V. Polshettiwar, B. Baruwati and R. S. Varma, Green Chem., 2009, 11, 127–131 RSC
- M. D. Le Page and B. R. James, Chem. Commun., 2000, 1647–1648 RSC
- Z. R. Dong, Y. Y. Li, S. L. Yu, G. S. Sun and J. X. Gao, Chin. Chem. Lett., 2012, 23, 533–536 CrossRef CAS
- A. O. Ogweno, S. O. Ojwach and M. P. Akerman, Dalton Trans., 2014, 43, 1228–1237 RSC
- A. A. Watson, D. A. House and P. J. Steel, Inorg. Chim. Acta, 1987, 130, 167–176 CrossRef CAS
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, 6th edn, 1999, p. 835 Search PubMed
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662–671 CrossRef CAS PubMed
- M. Taştekin, S. Durmuş, E. Şahin, C. Arici, K. C. Emregül and O. Atakol, Z. Kristallogr., 2008, 223, 424–429 CrossRef
- S. O. Ojwach, I. A. Guzei, L. L. Benade, S. F. Mapolie and J. Darkwa, Organometallics, 2009, 28, 2127–2133 CrossRef CAS
- R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC
- A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266–12280 CrossRef CAS PubMed
- K. Murata, T. Ikariya and R. Noyori, J. Org. Chem., 1999, 64, 2186–2187 CrossRef CAS
- R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS
- P. W. N. M. van Leeuwen, Appl. Catal., A, 2001, 212, 61–81 CrossRef CAS
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS
- P. Forzatti and L. Lietti, Catal. Today, 1999, 52, 165–181 CrossRef CAS
- M. Aydemir, N. Meric and A. Baysal, J. Organomet. Chem., 2012, 720, 38–45 CrossRef CAS
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 1046–1048 CrossRef CAS
- F. Calderazzo, U. Englert, C. Hu, F. Marchetti, G. Pampaloni, V. Passarelli, A. Romano and R. Santi, Inorg. Chim. Acta, 2003, 344, 197–206 CrossRef CAS
- I. A. Guzei and M. Wendt, Dalton Trans., 2006, 3991–3999 RSC
- R. Shannon and C. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 1046–1048 CrossRef CAS
- G. Seber and C. Wild, Nonlinear Regression, 1989, pp. 325–365 Search PubMed
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS PubMed
- A. Austin, G. A. Petersson, M. J. Frisch, F. J. Dobek, G. Scalmani and K. Throssell, J. Chem. Theory Comput., 2012, 8, 4989–5007 CrossRef CAS PubMed
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC
- S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS
- J. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS
- A. Landwehr, B. Dudle, T. Fox, O. Blacque and H. Berke, Chem.–Eur. J., 2012, 18, 5701–5714 CrossRef CAS PubMed
- D. L. Jameson and K. A. Goldsby, J. Org. Chem., 1990, 55, 4992–4994 CrossRef CAS
- APEX2, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed
- SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 Revisions C.01 and D.01 WIN64, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed
- A. McLean and G. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef
- P. Fuentealba, H. Preuss, H. Stoll and L. Von Szentpály, Chem. Phys. Lett., 1982, 89, 418–422 CrossRef CAS
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS
- OriginPro 9.1, OriginLab Corporation, One Roundhouse Plaza, Suite 303, Northampton, MA 01060, USA, 2013 Search PubMed
- C. Lo, J. Theor. Biol., 2007, 248, 317–321 CrossRef CAS PubMed
- X. Xu, Int. J. Bio-Med. Comput., 1987, 20, 35–39 CrossRef CAS PubMed
- M. M. Gil, T. R. Brandao and C. L. Silva, J. Food Eng., 2006, 76, 89–94 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1479994, 1479996, 1479997 and 1480100. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra12788f |
| ‡ Author contributions: M. N. M. and G. S. N. synthesized and characterized the compounds, performed the kinetics, and wrote parts of the Experimental method section and draft paper; S. O. O. directed the catalysis and co-wrote the manuscript; O. Q. M. performed the crystallography, data analysis, and DFT simulations, produced the figures, and co-wrote the manuscript. All authors have given their approval of the final version of the manuscript. |
| This journal is © The Royal Society of Chemistry 2016 |