
Preparation of piperlongumine-loaded chitosan nanoparticles for safe and efficient cancer therapy
 a
a
Abstract
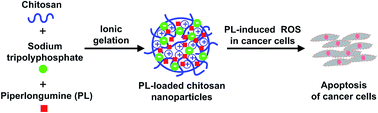
Development of biocompatible nanocarriers has gained much attention for cancer therapy because they can enhance cellular uptake and bioavailability of anticancer drugs with low toxicity. Chitosan has been intensively explored for biocompatible drug carriers due to high biodegradability and low toxicity. In this study, chitosan was used to synthesize biocompatible nanocarriers that can stably encapsulate piperlongumine (PL), a hydrophobic anticancer drug with cancer-specific antiproliferative activity via modulation of intracellular reactive oxygen species (ROS) in cancer cells. The PL-loaded chitosan nanoparticles (PL–CSNPs) prepared via ionic gelation showed an average particle size around 361 nm. The PL loading efficiency of the PL–CSNPs was 12 ± 2%. A drug release study revealed that the release of PL from the CSNPs was sustainable and pH-dependent. The PL–CSNPs showed efficient cytotoxicity against human gastric carcinoma (AGS) cells via dramatic increase of intracellular ROS leading to cell apoptosis. This study demonstrates that CSNPs are promising drug carriers for safe and effective PL delivery that can mediate efficient anticancer activity against gastric cancer cells.
 Please wait while we load your content...
Please wait while we load your content...

