
Molecular modeling of structural and functional variance in the SAGA deubiquitinating module caused by Sgf73 Y57A mutation†
Ya-Jyun Chen and
Chia-Ning Yang*
Department of Life Sciences, National University of Kaohsiung, Kaohsiung, Taiwan. E-mail: cnyang@nuk.edu.tw; Fax: +886-7-5919404; Tel: +886-7-5919717
First published on 30th June 2016
Abstract
The Spt-Ada-Gcn5-acetyltransferase deubiquitinating module is composed of four protein subunits: Ubp8, Sgf11, Sus1, and Sgf73. Recent biomolecular data has suggested that a mutation of Sgf73 wherein tyrosine-57 (Y57) is replaced by a smaller alanine residue introduces structural instability to the entire module, leading to a loss of deubiquitinating function. Notably, although Sgf73 Y57 is not directly involved in ubiquitin substrate binding, it finely tunes the structure and dynamics near the active site through intraprotein communication pathways. Here, we assessed possible allosteric mechanisms caused by Sgf75 Y57A mutation through molecular modeling. The data obtained suggest that such a mutation has a direct impact on Ubp8's fingers subdomain, which harbors the ubiquitin substrate's globular portion, and has an indirect influence on Sgf11's N-terminal helix, whose instability gradually perturbs Sgf11's C-terminal ZnF domain and Ubp8's Cys-His-Asn catalytic triad.
1. Introduction
Protein ubiquitination plays an essential role in regulating numerous cellular processes, including gene transcription, signal transduction, cell-cycle progression, protein trafficking, DNA repair, and DNA replication.1–4 Ubiquitination is regulated by ubiquitin deconjugation through deubiquitinating enzymes (DUBs), which are a special class of isopeptidases. The function of DUBs is to hydrolyze the conjugated adduct of ubiquitin (or ubiquitin-like protein) and a target protein joined by ubiquitin's Gly–Gly motif and the Lys of the target protein.5–7 Ubiquitin is a 76-amino-acid protein in a compact globular β-grasp fold followed by an extended tail where the Gly–Gly motif resides.8–11 The mechanism of DUB cysteine proteases is achieved by a Cys-His-Asp/Asn catalytic triad. Hydrolysis is initiated by a nucleophilic attack on the substrate carbonyl carbon by the anionic sulfur of the cystine, which is deprotonated by a nearby histidine residue. This histidine is stabilized by an asparagine or an aspartic residue. After the substrate releases a fragment with an amine terminus, the histidine residue returns to its deprotonated status while a thioester intermediate linking the new carbonyl terminus of the substrate is produced. The thioester bond is then hydrolyzed, generating a carboxylic acid terminus on the substrate and restoring the cysteine in the free cysteine proteases.12The Spt-Ada-Gcn5-acetyltransferase (SAGA) complex, which is composed of 21 proteins, is a 1.8 MDa transcriptional coactivator with two well-known functions, namely histone acetylation and deubiquitination of histone H2B.13–16 Because SAGA is structurally and functionally conserved from yeast to mammals, it has been a model for evaluating eukaryotic gene activation.17–19 The deubiquitination function of SAGA is achieved by the DUB module (DUBm), which consists of the ubiquitin-specific protease Ubp8 in a complex with Sgf11, Sus1, and Sgf73. With the exception of Sgf73, which participates in this module with only its N-terminal fragment (amino acid 1–96 or 1–104); all other subunits within the DUBm are full length.20 Fig. 1(a) depicts the SAGA DUBm bound with ubiquitin (Protein Data Bank [PDB] code: 3MHS), where the entire module can be bisected into the assembly lobe and the catalytic lobe. The assembly lobe is formed by the C-terminal of Ubp8, or the so-called Ubp8 zinc-finger ubiquitin binding (ZnF-UBP) domain, in contact with the other three subunits. The N-terminal helix of Sgf11 runs through the central region of the assembly lobe from the bottom to the top; at the end of this long helix is a loop followed by a ZnF domain that reaches the catalytic lobe. Sgf73 spreads over the assembly lobe and the lobe–lobe interface, whereas Sus1 functions as a clamp to unite the Sgf11, Sgf73, and Ubp8 in the assembly lobe.
 | ||
| Fig. 1 Overall view of yeast SAGA DUBm structure. (a) Reproduced view (based on PDB: 3MHS) of the SAGA DUBm in a complex with Ubal (in yellow). The whole module, classified as the assembly lobe and the catalytic lobe, comprises four protein units, namely Ubp8 (green), Sgf11 (red), Sgf73 (gold), and Sus1 (blue). Zn ions chelated by ZnF domains spread throughout the module are in pink. The catalytic site, an essential ZnF domain on Sgf11, and the secondary structures centered at the studied point mutation site at Sgf73 Y57 are specified. (b) Lateral view of the catalytic lobe comprising the fingers (yellow-green), thumb (pale green), and palm (lime green) subdomains of Ubp8 and the N-terminal ZnF domain (red) of Sgf11. From bottom to top, the catalytic lobe holds the globular portion of Ubal and its C-terminal tail, whose G76, here capped by an aldehyde group to avoid hydrolysis, brings the segment to be hydrolyzed into the catalytic triad. The specified steps 1–3 are for discussion purposes. (c) Catalytic triad composed of C146, H427, and N443 in Ubp8. (d) The Ubal C-terminal tail passes through a slender tunnel encompassed by the Ubp8 thumb (pale green) and palm (lime green) subdomains and interacts with Ubp8 loop L1 (black). (e) Numerous electrostatic interactions toward the Ubal globular portion from the Ubp8 fingers and palm subdomains. (f) Electrostatic surface potentials of the Ubp8's fingers domain and the Ubal's globular portion to show their paired electrostatic interactions linked by dotted lines. | ||
The catalytic lobe is mainly composed of the Ubp8 catalytic domain capped by the aforementioned Sgf11 ZnF domain for stabilizing the Ubp8 catalytic site boxed in Fig. 1(a). As with other DUBs, the Ubp8 catalytic domain is divided into three subdomains: fingers, palm, and thumb subdomains,11 as indicated in the lateral view of the catalytic lobe depicted in Fig. 1(b). The fingers subdomain is responsible for holding the globular portion of ubiquitin with the hydrophobic interactions and hydrogen bonds highlighted in Fig. 1(e). As shown in Fig. 1(b), between the thumb and palm subdomains is a narrow tunnel that accommodates the extended C-terminal tail of ubiquitin, which reaches the cysteine protease active site for hydrolysis. In Fig. 1(c), catalytic triad comprises C146, H427, and N443, where the nearby N141 is proposed to stabilize the oxyanion intermediate.21 According to a structural comparison between the SAGA DUBm–ubiquitin aldehyde (Ubal) complex (PDB code: 3MHS) and apo SAGA DUBm (PDB code: 3MHH), Wolberger et al. indicated that such a catalytic triad in the apo form adopts a catalytically competent arrangement where C146 is ready for deprotonation by H427, whose imidazole moiety is stabilized by N443.20
Although Ubp8 maintains a catalytic domain similar to those of the ubiquitin-specific protease family of deubiquitinating enzymes, it is inactive without being complexed with Sgf11, Sus1, and Sgf73. Recently several studies have revealed the role played by Sgf73 and Sgf11 in maintaining SAGA DUBm structure and function.22–25 Wolberger's group speculated that Sgf73 Y57, N59, and N61 (the first, third, and fifth amino acid residues on loop L3 shown in Fig. 1(a)), may provide backside support for the Ubp8 fingers subdomain to hold the globular portion of ubiquitin. Activity assay experiments showed that the Sgf73 Y57A mutant abrogates deubiquitinating activity completely, whereas N59D and N61D mutants introduce moderate vulnerability to the whole module.19 Wolberger and colleagues also determined the crystal structures of Y57A and N59D and noted that, in both mutants, the C-terminal ZnF of Sgf11 that caps the catalytic site is partially disordered in N59D mutant and completely disordered in Y57A mutant. Notably, Sgf73 Y57 and N59 residues are remote from Sgf11 ZnF, but they indirectly perturb the stability associated with Sgf11 and Ubp8. Further analysis on the Y57A mutant demonstrated its deubiquitinating activity through a gel assay, and its binding affinity toward diubiquitin was determined to be temperature sensitive through isothermal titration calorimetry (ITC).19 The gel assay showed that Y57A mutant remains slightly active at temperatures between 10 and 25 °C but is completely inactive at 35 °C. In the ITC experiment, the dissociation constant (Kd) for the wild type (WT, where the catalytic C146 of Ubp8 was replaced by alanine to prevent hydrolysis) toward diubiquitin at 24 °C was 0.4 μM, whereas the Kd values for Y57A mutant (also with Ubp8 C146A replacement) measured at 20, 24, 28, and 32 °C were 0.4, 0.67, 1.2, and 6.7 μM, respectively, with Kd at 35 °C unable to be determined. The ITC data indicating the ability of Y57A mutant to bind to ubiquitin at low temperature but not at high temperature implies a certain degree of dynamic variance caused by mutating Y57 to the smaller A57 residue.
Prior to our molecular dynamics (MD) simulations, we evaluated the apo and ubiquitin-bound SAGA DUBm structures with PDB codes 3MHH and 3MHS, respectively, and proposed three steps, which are illustrated in Fig. 1(b), for SAGA DUBm–ubiquitin recognition to achieve the proteolytic reaction. The first step is accommodation of the ubiquitin globular portion by the Ubp8 fingers and thumb subdomains as shown in Fig. 1(e), a rotated view of the lower part of Fig. 1(b). This component of Ubp8–ubiquitin interaction involves several hydrogen bonds, salt bridges, and van der Waals contacts. Hydrogen bond interactions include K363-T7′/L8′ and E327-T12′ in the Ubp8–ubiquitin interface, whereas salt bridges include K297-E64′ and D331-K33′. K363 is located on the thumb subdomain, whereas E327, K297, and D331 are all found on the fingers subdomain. Therefore, the collective movement of the fingers and thumb subdomains is an essential factor in guiding the protein–protein recognition precisely, as shown in the electrostatic potential surfaces constructed for the Ubp8–ubiquitin interface in Fig. 1(f). Fig. 1(b) indicates that in the second step, the ubiquitin C-terminal tail passes through a slender tunnel surrounded by the Ubp8 thumb and palm subdomains and interacts with Ubp8 loop L1 (shown in black). Structural analysis shows Q236/Q235-R74′ and D237-L73′ hydrogen bonds between Ubp8 L1 and the ubiquitin C-terminal tail of the SAGA DUBm–ubiquitin complex (Fig. 1(d)), whereas loop L1 was too flexible to be determined in the apo SAGA DUBm (PDB code: 3MHH). The dissimilar behaviors of Ubp8 loop L1 indicates that it undergoes a conformational change driven by induced fit to align with the incoming ubiquitin C-terminal tail. In step 3, the very end of the ubiquitin C-terminal tail interacts with the Cys-His-Asn catalytic triad so that the deubiquitinating reaction proceeds (Fig. 1(c)). This step requires a proper arrangement of C146, H427, and N443 residues in the Ubp8 active site.
MD simulations have long been recognized as a powerful modeling tool in providing structural and dynamic details at atomic level to interpret the available experimental observations.26–30 For example, previously we applied MD simulations for the unbound forms of Keap1WT and six Keap1MTs and provided rationale to correlate to the experimentally observed two mutated sites not involved in the Keap1–Nrf2 interface that can severely abolish the protein–protein recognition whereas four mutated sites on the interface still maintaining considerable Keap1–Nrf2 affinity.26 On the other hand, MD simulation approach such as alanine scanning can play an auxiliary role in predicting hot spots of protein–protein interaction that serve as useful information for biomolecular experimentalists in designing new drug compounds.31–33 In this study, we applied MD simulations to the apo WT and Y57A mutant to interpret the allosteric impact on SAGA DUBm function and structure variance introduced by replacing Sgf73 Y57 with a smaller alanine residue.
2. Experimental methods
Currently, three crystal structures of the Saccharomyces cerevisiae SAGA DUBm are available in the PDB: 3MHH (apo WT), 4W4U (apo Y57A mutant), and 3MHS (WT bound with Ubal).14,19 Here, MD simulations on the apo WT and apo Y57A mutant of the SAGA DUBm were performed to discover possible structural and dynamic discrepancies responsible for varied enzymatic activity. The Ubal-bound structure served as a structural reference. In these two simulated SAGA DUBm systems, the N- and C-termini were capped by acetyl and N-methyl groups, respectively. Several missing portions of the WT and the Y57A mutant proteins were repaired and optimized on the basis of the SAGA DUBm–Ubal complex structure, with the exception of the undetermined Sgf11 ZnF domain on top of the catalytic lobe of the Y57A mutant. The PDB file (4W4U) shows the loop linking this ZnF domain, and the long helix running through the assembly lobe is highly dynamic. Therefore, according to our experience with MD simulations, positioning the Sgf11 ZnF domain back on the top of the catalytic lobe in the Y57A mutant could introduce bias to the dynamic and structural analyses. All missing portions added to the studied systems are represented as black fragments in Fig. S1(a) and (b)† for the WT and Y57A mutant, respectively. Each studied system was immersed in a cubic box of the TIP3P water model34,35 with an appropriately sized box, which resulted in distances between the atoms in each SAGA DUBm and the wall that were greater than 12 Å. To neutralize the studied systems, 24 K+ ions and 22 K+ ions were added to the WT and Y57A, respectively. To enrich the abundance of simulation data, we submitted three independent simulation runs for each of WT and Y57A mutant.The neighboring TIP3P water molecules within 1.5 Å of the complex were removed to prevent biased hydrogen bonds between the studied SAGA DUBm and the added solvent molecules at the beginning of the simulation. Each solvated system was energy minimized by considering three stages, each employing 500 steps of the steepest descent algorithm and 500 steps of the conjugate gradient algorithm with a nonbonded cutoff of 8.0 Å. At stage one, the SAGA DUBm structure was restrained so that the added TIP3P water molecules reoriented properly. At stage two, the backbone of the SAGA DUBm was restrained so that the amino acid side chains could find appropriate ways to avoid neighboring specious conflict. At stage three, the entire solvated system was minimized without any restraint.
We conducted MD simulations by using the AMBER 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36,37 software package. The cationic dummy atom approach38 and the AMBER FF99SB force field39,40 were employed to treat the ZnF motif and the rest of the system, respectively. All MD simulations of the two systems were performed according to the standard protocol, which entails gradual heating, density, equilibration, and production procedures in the isothermal isobaric ensemble (NPT, P = 1 atm and T = target temperature) MD. A minimized solvated system was used as the starting structure for subsequent MD simulations. In the 500 ps heating procedure, the system was gradually heated from 0 to the target temperature in 50 ps, followed by density at the target temperature 303 K for 500 ps and constant equilibration at the target temperature 303 K for 50 ps. After the equilibration procedure, the system underwent a 30 ns production procedure for conformation collection. The time step was set to 2 fs. A snapshot was captured every 10 ps to record the conformation trajectory during production MD. An 8 Å cutoff was applied to treat nonbonding interactions, such as short-range electrostatic and van der Waals interactions, whereas the particle mesh Ewald method41 was applied to treat long-range electrostatic interactions. The SHAKE algorithm42,43 was used to constrain all bonds containing hydrogen atoms to their equilibrium lengths. Equations to estimate RMSD, RMSF, and PCA are given in ESI† part.
36,37 software package. The cationic dummy atom approach38 and the AMBER FF99SB force field39,40 were employed to treat the ZnF motif and the rest of the system, respectively. All MD simulations of the two systems were performed according to the standard protocol, which entails gradual heating, density, equilibration, and production procedures in the isothermal isobaric ensemble (NPT, P = 1 atm and T = target temperature) MD. A minimized solvated system was used as the starting structure for subsequent MD simulations. In the 500 ps heating procedure, the system was gradually heated from 0 to the target temperature in 50 ps, followed by density at the target temperature 303 K for 500 ps and constant equilibration at the target temperature 303 K for 50 ps. After the equilibration procedure, the system underwent a 30 ns production procedure for conformation collection. The time step was set to 2 fs. A snapshot was captured every 10 ps to record the conformation trajectory during production MD. An 8 Å cutoff was applied to treat nonbonding interactions, such as short-range electrostatic and van der Waals interactions, whereas the particle mesh Ewald method41 was applied to treat long-range electrostatic interactions. The SHAKE algorithm42,43 was used to constrain all bonds containing hydrogen atoms to their equilibrium lengths. Equations to estimate RMSD, RMSF, and PCA are given in ESI† part.
3. Results and discussion
3.1 MD stability of WT and Y57A mutant
To evaluate MD trajectory quality and convergence, the Cα root-mean-square deviation (RMSD) values for the WT and the Y57A mutant in the production duration as a function of time were plotted in Fig. 2. The curves in Fig. 2(a) for the WT and in Fig. 2(b) suggest that the three simulated WT structures and three simulated Y57A mutant structures remained stable for nearly the entire MD duration. | ||
| Fig. 2 Time evolution of the Cα RMSD of the SAGA DUBm: (a) for the three simulation runs of WT, (b) for the three simulation runs of Y57A mutant. | ||
For WT and Y57A mutant, structural and dynamic analysis were performed based on the 300 conformations by gathering 100 snapshots with equal time intervals between 20 and 30 ns in each of the three simulation runs for either WT or Y57a mutant. With the collected 300 conformations, the root mean square fluctuation (RMSF) per amino acid residue in each of the four components of the SAGA DUBm was also calculated and plotted in Fig. 3. In Fig. 3(a), the Ubp8 of the WT has a flexible fingertip and a loop behind the fingers, which is hereafter referred to as the 388–408 loop, according to the beginning and ending amino acid numbers. This is in line with the apo WT X-ray crystallography structure for these two regions, which were too flexible to be determined. The Ubp8 of Y57A contains more regions with high mobility, which are found in the palm, fingertip, and the 338–408 loop; however, the 338–408 loop was not solved in the X-ray crystallography structure. The Ubp8 catalytic domain in both the WT and Y57A mutant is generally dynamic in comparison with the static ZnF-UBP domain intertwined with Sus1, Sgf11, and Sgf73 in the assembly lobe. In Fig. 3(b), showing Sgf73, the fragment beyond K55 is sandwiched between the assembly lobe and the catalytic lobe and consequently has low RMSF values estimated in both the WT and Y57A. The fragment preceding Q45 shows higher RMSF for this part and is located on the assembly lobe surface or is exposed in the solvent, as indicated in Fig. 1(a). Fig. 3(c) shows that the Sgf11 N-terminal ZnF in the WT possesses high RMSF values, whereas in Y57A mutant the coordinates of this section were too flexible to be determined and were not included in our simulation data. In both the WT and the Y57A mutant, the long helix embraced mainly by Sus1 and Ubp8 and marginally by Sgf73 is rigid. Because the C-terminal ZnF domain was not solved in Y57A mutant, the long loop between ZnF and the long helix is mobile in Y57A mutant. Because Sus1 is embedded in the assembly lobe, its RMSF estimated for both WT and Y57A mutant is relatively low in Fig. 3(d).
 | ||
| Fig. 3 Estimated RMSF per residue according to the conformations collected from the last 10 ns in each of the three WT simulations and the three Y57A simulations: (a) Ubp8, (b) Sgf73, (c) Sgf11, and (d) Sus1. | ||
3.2 Dynamics and structure variance of the fingers subdomain
On the basis of the aforementioned three steps proposed for the recognition process between SAGA DUBm and ubiquitin, we conducted structural and dynamic analyses to determine how the mutation on Sgf73 Y57A contributes to the loss of DUBm function. Principal component analysis (PCA) of the MD trajectories was conducted to characterize the essential dynamic difference related to the reduced SAGA DUBm–ubiquitin binding affinity of Y57A mutant (Kd = 0.67 μM at 24 °C and not determinable at 35 °C in Y57A mutant, and Kd = 0.4 μM at 24 °C in Wolberger's ITC experiment on the WT).26 The first PCA modes were found to account for 73% and 68% of the total dynamic variance for the WT and Y57A mutant, respectively. Fig. 4(a) and (b) depict the dynamics of the WT and Y57A expressed by their first PCA modes with RMSF found in the palm, fingertip, and the 388–408 loop, which is consistent with the highly flexible regions in the Ubp8 RMSF plot (Fig. 3(a)). In Fig. 4(a), the WT fingers subdomain moves relative to the palm subdomain in the same direction, whereas in Fig. 4(b) the movements of these two subdomains in Y57A mutant are not parallel. More specifically, in the WT, the palm and fingers move simultaneously downward and slightly to the left-hand side, whereas in Y57A mutant the palm moves downward and slightly to the right-hand side while the fingers move to the left-hand side. The advantage of the parallel movement of the fingers and palm in the WT are twofold: first, it is more energetically favourable to maintain a relatively steady and constant pocket volume to accommodate the globular portion of the ubiquitin substrate. Moreover, as the relative positions of K363 in the Ubp8 palm subdomain and K297, E327, and D331 in the Ubp8 fingers subdomain pinpointed in Fig. 1(f) are confined, the protein–protein recognition between Ubp8 and the ubiquitin substrate through long-range electrostatic interaction is efficient. | ||
| Fig. 4 Dynamic and structural analyses of the Ubp8 fingers subdomain. (a) PC1 data for the WT, where the color code for high to low mobility ranges from red to white to blue. (b) PC1 for Y57A mutant, in the same color code as in WT. (c) Structural details on the Sgf73 H3-L3 junction, Ubp8 fingers subdomain, and 388–408 loop in the WT. The hydrogen bond occupancies are 92% for Y57-T394, 100% for Y57-L354, 89% for Y391-I351, and 83% for S393-I351. (d) Structural details on the Sgf73 H3-L3 junction, Ubp8 fingers subdomain, and 388–408 loop in Y57A mutant. The hydrogen bond occupancies are 100% for A57-L354, 88% for Y391-I351, 89% for S393-I351, and 44% for T394-E284. | ||
Structural analysis was further performed to correlate Sgf73 Y57/A57 with the Ubp8 dynamic behaviours in the WT and Y57A mutant. Fig. 4(c) shows that in the WT, a hydrogen bond network joins Y57 (the first amino acid residue on Sgf73 loop L3); Y391, S393, and T394 (on the 388–408 loop); and I351 and L354 (on the β7 of fingers). Likewise, the Y57A mutant in Fig. 4(d) holds a hydrogen bond network to bring A57 (the mutated residue that replaces Sgf73 Y57), Y391, C392, and S393 (on the 388–408 loop); I351 and L354 (on the β7 of fingers); and E284 (on the β1 of fingers) in close contact. In the WT, because Sgf73 Y57 uses its phenyl hydrogen atom to form a hydrogen bond with T394 as well as its bulky residue surface to provide van der Waals contact with other amino acid residues on the 388–408 loop, the movement of this loop is slower than it is in the Y57A mutant where the smaller A57 is not able to access residues beyond Y391. The mutated A57 reduces the interaction between Sgf73 helix H3 and the 388–408 loop, which is evidenced by the hydrogen bond network in Fig. 4(d). This is consistent with the increased loop mobility represented by PCA in Fig. 4(b) and by the RMSF estimated per amino acid in Fig. 3(a).
In addition, Wolberger et al. indicated that Sgf73 Y57A mutant causes an inward movement of the fingers subdomain that further shrinks the pocket for the ubiquitin globular portion and hampers the protein–protein recognition between the SAGA DUBm and ubiquitin. This is based on structural alignment in reference to the thumb and palm of Ubp8 for the apo SAGA DUBmY57A and apo SAGA DUBmN59D (where N59 is also located on Sgf73) showing a shorter palm–fingers separation than that of the apo SAGA DUBmWT.26 Our structural analysis in Fig. 4(d) suggests that two firm hydrogen bonds between E284 and S393 are exclusively found in Y57A mutant, accounting for the inward movement of the fingers subdomain β-sheet. Shown in Fig. S2(a)† is a reproduced structural alignment of the Ubal-bound SAGA DUBmWT, apo SAGA DUBmWT, and apo SAGA DUBmY57A structures solved by Wolberger et al. We discovered that in the apo SAGA DUBmY57A, the linkage between E284 and S393 pulls β4 and β7 is very close to the 388–408 loop (or outward from the ubiquitin binding side) so that β1 and β2 are pushed away from the 388–408 loop (or inward toward the ubiquitin binding side), as indicated in Fig. S2(d).† For comparison, refer to the structure of the SAGA DUBm–Ubal complex in Fig. S2(b)† and that of the SAGA DUBmWT in Fig. S2(c).† Especially for the upward movement of the β2 strand, K297 moves upward and shortens the K297-D331 separation distance (13.6 Å for the Y57A mutant vs. 16.8 Å for the WT from K297 Cα to D331 Cα). The inward movement of the fingers in the Y57A mutant rearranges the relative positions of K297, E327, D331, and K363, which play essential roles in recognizing the globular portion of ubiquitin.
3.3 L1 conformational change upon ubiquitin binding
In Fig. 5(a)–(c), a comparison among the X-ray crystallography determined structures of SAGA DUBmWT–Ubal complex, SAGA DUBmWT, and SAGA DUBmY57A reveals an obvious discrepancy on loop L1 (from N227 to D237). In the SAGA DUBmWT–Ubal complex, numerous hydrogen bonds formed from Q236 and D237 on Ubp8 loop L1 toward L73′ and R74′ on the ubiquitin C-terminal tail, as highlighted in Fig. 1(d), play an essential role in stabilizing the ubiquitin C-terminal tail and further guide it toward the catalytic triad for hydrolysis. Regardless of the structural importance of loop L1, in both the SAGA DUBmWT and SAGA DUBmY57A the segment between Q228 and Y233 is disordered and not determined, and the solved Q235-Q236-D237 segment is oriented away from the slender tunnel to accommodate the ubiquitin C-terminal tail, as shown in Fig. 5(b) and (c). Notably, a groove to properly place loop L1 upon ubiquitin binding exists in the SAGA DUBmWT (Fig. 5(d)) implying that the highly flexible loop L1 undergoes an induced-fit-based conformational change in the presence of a ubiquitin substrate. Moreover, our structural analysis of the SAGA DUBmWT–Ubal complex indicates that the slender tunnel for harbouring the ubiquitin C-terminal tail is roofed by Q235 (on loop L1) and E425 (on the palm), as delineated in Fig. 5(d). This is consistent with the observed high flexibility of loop L1, which is based on the undetermined coordinates of loop L1 in the apo SAGA DUBmWT X-ray crystallography solved structure. Contact between Q235 and E425 is avoided, thereby making the slender tunnel more accessible. In our MD simulations for WT, we constructed loop L1 on the basis of the solved Q228-Y233 segment in the SAGA DUBmWT–Ubal complex structure and the solved Q235-D237 segment in the apo SAGA DUBmWT structure. Although the repaired loop L1 does not show high flexibility in its RMSF relative to other parts of Ubp8 plotted in Fig. 3(a), within the entire MD simulation duration loop L1 does not move toward the position discovered in the SAGA DUBmWT–Ubal complex structure. Shown in Fig. 5(e) is a snapshot taken from the very beginning of WT simulation where the repaired loop L1 fits into the existent groove. | ||
| Fig. 5 Structural comparison among the three X-ray crystallography determined SAGA DUBm structures for loop L1. (a) Ubal-bound SAGA DUBm complex (PDB code: 3MHS) where loop L1 (N227-D237) was completely determined. (b) Apo form of the SAGA DUBmWT (PDB code: 3MHH) where Q228-Y233 was not determined and the solved fragment between S234 and D237 is oriented away from the Ubal-bound form. (c) Apo form of the SAGA DUBmY57A (PDB code: 4W4U) where N227-Q235 was not determined and the solved fragment between Q236 and D237 is oriented away from the Ubal-bound form. (d) In the Ubal-bound SAGA DUBm complex, loop L1 fits into a groove. The bulky residue of W224 is oriented upward because of van der Waals contact with the Sgf11 ZnF domain. (e) The apo form of the SAGA DUBmWT has a groove to place the undetermined loop L1 when loop L1 is reconstructed on the basis of the Ubal-bound SAGA DUBm complex. (f) In the apo form of the SAGA DUBmY57A, a groove to place the undetermined loop L1, based on the Ubal-bound SAGA DUBm complex, does not exist. Here, W224 flips down to fill the groove for loop L1. | ||
As for the SAGA DUBmY57A structure (PDB: 4W4U) whose loop L1 was not determined, the groove for properly positioning L1 is not present, as illustrated in Fig. 5(f). Its absence implies some inherent obstacle for loop L1 undergoing a conformational change upon ubiquitin binding. Our structural analysis on apo SAGA DUBmY57A suggests that the bulky W224 fills the groove for loop L1 (Fig. 5(f)); whereas in the WT, W224 interacts with Sgf11 and is away from the groove (Fig. 5(e)).
3.4 Disorder of the catalytic triad
Fig. 6 shows the diverse catalytic triad arrangements in the SAGA DUBmWT and SAGA DUBmY57A. As illustrated in Fig. 6(a), the spatial settlement of the catalytic triad (C146, H427, and N443) greatly relies on the Sgf11 ZnF. Sgf11 K63 anchors Ubp8 D444, propagating structural stability for D443 and H427. The hydrogen bond between Sgf11 A86 and Ubp8 M142 secures loop L2 so that C146 is oriented in a manner ready for deprotonation by H427 and N141 in an appropriate position to stabilize the oxyanion intermediate. As for the SAGA DUBmY57A, whose Sgf11 ZnF coordinates are too flexible to be determined in the X-ray crystallography structure, neither Ubp8 L2 nor D444 is secured by Sgf11. Fig. 6(b) shows that hydrogen bond linkages formed between N443 and G54 (Sgf11) and between C146 and D444 leave H427 to be stabilized by its neighboring Y428. Accordingly, once the ubiquitin substrate is bound to Y57A, the catalytic site must rearrange C146, H427, and N443 so that proteolysis can proceed. However, without the structural anchoring aided by the ZnF domain of Sgf11, the rearrangement would occur with difficulty. | ||
| Fig. 6 Arrangement of the catalytic triad in (a) the WT and (b) Y57A mutant. | ||
3.5 Correlating A57 to the missing Sgf11 ZnF in Y57A mutant
The Sgf11 C-terminal ZnF domain is essential to the deubiquitinating activity in the SAGA DUBm, because it facilitates maintaining the catalytic triad arrangement and both dynamic and structural guidance for loop L1's induced-fit and further interaction with the C-terminal tail of the incoming ubiquitin substrate. As shown in Fig. 1(a), Sgf11 begins with a long helix running through the assembly lobe, followed by a flexible long loop flanking behind the top region of the assembly and catalytic lobes, and ends with a ZnF domain capping the Ubp8 catalytic site. In the X-ray crystallography-solved SAGA DUBmY57A structure, the coordinates of the Sgf11 ZnF domain were not determined owing to its structurally disordered nature, whereas the same region in the ubiquitin-bound SAGA DUBmWT and apo SAGA DUBmWT structures was determined. Because the long loop of Sgf11 linking the N-terminal helix and the C-terminal ZnF domain is also flexible and possesses very few contacts with Ubp8, we focused on the long helix surrounded by Ubp8, Sus1, and Sgf73 in the assembly lobe to correlate the missing ZnF domain in SAGA DUBmY57A. Table S1† lists a hydrogen bond survey of the collected trajectories for the WT and Y57A MD simulations, alongside the ubiquitin-bound SAGA DUBmWT complex structure for comparison. In the WT column, 11 stable hydrogen bonds, mainly from Ubp8 and partly from Sgf73, provide stability for the long helix of Sgf11. Notably, although the hydrogen bond occupancy is 70% to 74% between Sgf11 R30 and Sgf73 D70 and between Sgf11 R41 and Sgf73 Q74, each amino acid residue pair comes with two hydrogen bonds that might coexist at some points in time but not in others. In the Y57A column, 11 hydrogen bonds were also observed but were found to have low occupancy, especially those between Sgf11 and Sgf74 in comparison with the WT. We plotted these hydrogen bond interactions in two formats in Fig. 7 to provide different points of view. Fig. 7(a) and (b) shows side views for the WT and Y57A, respectively, whereas Fig. 7(c) and (d) depict top views for the WT and Y57A mutant, respectively. Fig. 7(a) shows that in the WT, approximately two thirds (from T22 to R41) of the long helix of Sgf11 (from I6 to R41) are stabilized by strong hydrogen bonds from Ubp8 and Sgf73. Moreover, from the bottom to top of the helix, the hydrogen bond interaction contributors originate from Ubp8 and gradually from Sgf73. Meanwhile, the interaction originates from the lower left and gradually from the upper right. In Fig. 7(b), showing Y57A mutant, only the upper half the helix is secured by several weak hydrogen bonds whose occupancies are less than 60%. Three strong hydrogen bonds occur in the middle of the helix to secure Q25 and D26. A comparison between Fig. 7(a) and (b) suggests that in the WT, the top of this long helix leans to the right hand side, which may help the rest of the Sgf11 structure propagate toward the catalytic lobe. By contrast, the upper half of this long helix maintains weak interaction with Ubp8, Sgf73, and Sus1, showing no preferential orientation for the structure after the long helix, and therefore it would be less efficient for the ZnF domain to reach the top of the Ubp8 catalytic site. Fig. 7(c) and (d) shows the hydrogen bond partners in a radial distribution approach, which indicates that the R30-D70 and R41-Q74 interactions in the WT are crucial in orienting the long loop between the ZnF domain and the long helix from the assembly lobe toward the catalytic lobe. | ||
| Fig. 7 Stability of the long helix of Sgf11 conferred by the surrounding subunits in the two studied systems: (a) a side view of WT, (b) a side view of Y57A, (c) a top view of the WT, and (d) a top view of Y57A. Dash and solid arrows represent weak and steady hydrogen bonds with occupancies lower than or higher than 40%. Green, brown, and blue arrows are hydrogen bonds from Ubp8, Sgf73, and Sus1, respectively. | ||
4. Conclusions
Fig. 8 was generated to summarize the structural and dynamic observations regarding the loss of the SAGA DUBm caused by replacing the Sgf73 Y57 residue with a smaller alanine. The impact is twofold: first, the mutated A57 enhances the hydrogen bond network, joining Sgf73 helix H3 and the Ubp8 fingers domain and the 388–408 loop behind the fingers subdomain. That is, in the WT, the interaction between the 388–408 loop and the fingers subdomain (comprising β4, β7, β1, and β2 strands) is relatively minor in comparison with that in Y57A, where S393 forms two hydrogen bonds with E284 on the β1 strand and Y391 forms two hydrogen bonds with I351 on the β7 strand. The E284-S393 hydrogen bonds rotate β1 and β2 upward, bring the β4–β1 loop and β1–β2 loop on the fingertip toward the palm subdomain, and consequently shrink the binding pocket for accommodating the ubiquitin globular portion. The second impact is indirect but crucial. The mutated A57 slightly alters the interaction between Sgf73 loop L3 and the assembly lobe-catalytic lobe interface, further diminishing the interaction between Sgf73 L3 and the long helix of Sgf11 embedded in the assembly lobe. Without the stability conferred by the four steady D70-R30 and D74-R41 hydrogen bonds between Sgf73 and Sgf11, in Y57A mutant, the C-terminal end of the Sgf11 long helix is not properly anchored and structural disorder propagates along the extended loop and gradually toward the ZnF domain in the Sgf11 C-terminal end. With the missing Sgf11 ZnF domain on the top of the Ubp8 catalytic site, the catalytic triad is disordered as well, and a groove for relocating loop L1 upon interaction with the ubiquitin C-terminal tail is blocked by a bulky W224 residue. In the WT, the ring moiety of W224 faces Sgf11 because of van der Waals interaction.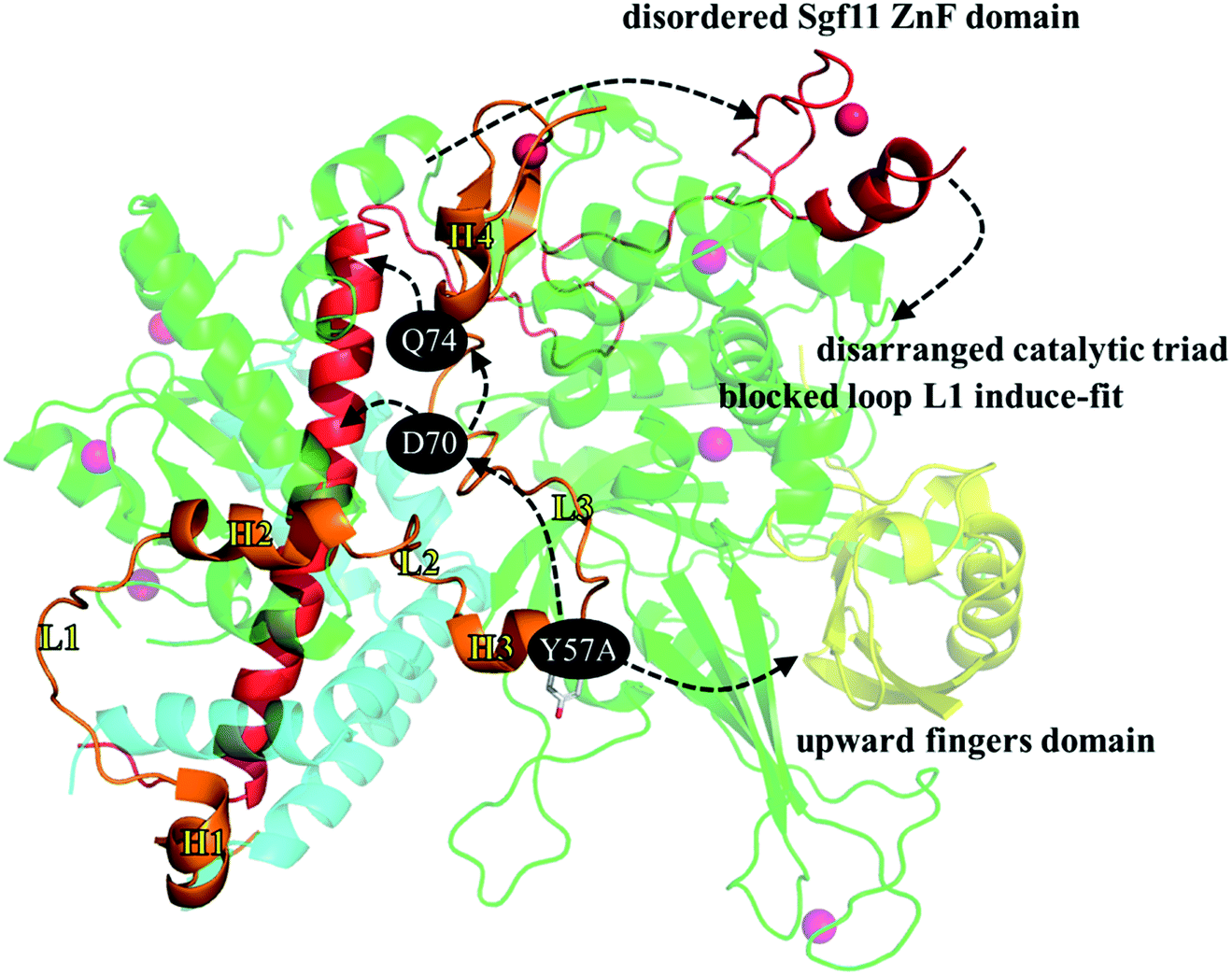 | ||
| Fig. 8 Summary of the structural and dynamic impact caused by replacing the Sgf73 Y57 residue with a smaller alanine. | ||
In summary, we began with a structural analysis of the available X-ray crystallography structures of the ubiquitin-bound SAGA DUBm, SAGA DUBmWT, and SAGA DUBmY57A and further applied MD simulations to explore the dynamics of the SAGA DUBmWT and SAGA DUBmY57A. Using these methods, we gained a clearer understanding of how substituting Sgf73 Y57 with an alanine influences the remote Ubp8 catalytic site through allosteric regulation.
Acknowledgements
The authors gratefully acknowledge the financial support provided for this study by the Ministry of Science and Technology of Taiwan (MOST 104-2113-M-390-003).Notes and references
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS PubMed.
- A. F. Alpi, P. E. Pace, M. M. Babu and K. J. Patel, Mol. Cell, 2008, 32, 767–777 CrossRef CAS PubMed.
- C. Chen, A. K. Seth and A. E. Aplin, Mol. Cancer Res., 2006, 4, 695–707 CrossRef CAS PubMed.
- H.-G. Zhang, J. Wang, X. Yang, H.-C. Hsu and J. D. Mountz, Oncogene, 2004, 23, 2009–2015 CrossRef CAS PubMed.
- A. Y. Amerik and M. Hochstrasser, Biochim. Biophys. Acta, Mol. Cell Res., 2004, 1695, 189–207 CrossRef CAS PubMed.
- Y. Sato, A. Yoshikawa, A. Yamagata, H. Mimura, M. Yamashita, K. Ookata, O. Nureki, K. Iwai, M. Komada and S. Fukai, Nature, 2008, 455, 358–362 CrossRef CAS PubMed.
- J.-F. Trempe, Curr. Opin. Struct. Biol., 2011, 21, 792–801 CrossRef CAS PubMed.
- D. Komander, F. Reyes-Turcu, J. D. Licchesi, P. Odenwaelder, K. D. Wilkinson and D. Barford, EMBO Rep., 2009, 10, 466–473 CrossRef CAS PubMed.
- F. E. Reyes-Turcu, J. R. Horton, J. E. Mullally, A. Heroux, X. Cheng and K. D. Wilkinson, Cell, 2006, 124, 1197–1208 CrossRef CAS PubMed.
- M. Hu, P. Li, L. Song, P. D. Jeffrey, T. A. Chernova, K. D. Wilkinson, R. E. Cohen and Y. Shi, EMBO J., 2005, 24, 3747–3756 CrossRef CAS PubMed.
- M. Hu, P. Li, M. Li, W. Li, T. Yao, J.-W. Wu, W. Gu, R. E. Cohen and Y. Shi, Cell, 2002, 111, 1041–1054 CrossRef CAS PubMed.
- G. Dodson and A. Wlodawer, Trends Biochem. Sci., 1998, 23, 347–352 CrossRef CAS PubMed.
- S. Rodríguez-Navarro, EMBO Rep., 2009, 10, 843–850 CrossRef PubMed.
- N. L. Samara, A. B. Datta, C. E. Berndsen, X. Zhang, T. Yao, R. E. Cohen and C. Wolberger, Science, 2010, 328, 1025–1029 CrossRef CAS PubMed.
- S. Rodríguez-Navarro, T. Fischer, M.-J. Luo, O. Antúnez, S. Brettschneider, J. Lechner, J. E. Pérez-Ortín, R. Reed and E. Hurt, Cell, 2004, 116, 75–86 CrossRef.
- K. W. Henry, A. Wyce, W.-S. Lo, L. J. Duggan, N. T. Emre, C.-F. Kao, L. Pillus, A. Shilatifard, M. A. Osley and S. L. Berger, Genes Dev., 2003, 17, 2648–2663 CrossRef CAS PubMed.
- T. Suganuma and J. L. Workman, Cell, 2008, 135, 604–607 CrossRef CAS PubMed.
- V. M. Weake and J. L. Workman, Trends Cell Biol., 2012, 22, 177–184 CrossRef CAS PubMed.
- M. Yan and C. Wolberger, J. Mol. Biol., 2015, 427, 1765–1778 CrossRef CAS PubMed.
- A. Köhler, E. Zimmerman, M. Schneider, E. Hurt and N. Zheng, Cell, 2010, 141, 606–617 CrossRef PubMed.
- N. L. Samara, A. E. Ringel and C. Wolberger, Structure, 2012, 20, 1414–1424 CrossRef CAS PubMed.
- K. Ingvarsdottir, N. J. Krogan, N. T. Emre, A. Wyce, N. J. Thompson, A. Emili, T. R. Hughes, J. F. Greenblatt and S. L. Berger, Mol. Cell. Biol., 2005, 25, 1162–1172 CrossRef CAS PubMed.
- K. K. Lee, L. Florens, S. K. Swanson, M. P. Washburn and J. L. Workman, Mol. Cell. Biol., 2005, 25, 1173–1182 CrossRef CAS PubMed.
- P. Pascual-García, C. K. Govind, E. Queralt, B. Cuenca-Bono, A. Llopis, S. Chavez, A. G. Hinnebusch and S. Rodríguez-Navarro, Genes Dev., 2008, 22, 2811–2822 CrossRef PubMed.
- K. K. Lee, S. K. Swanson, L. Florens, M. P. Washburn and J. L. Workman, Epigenet. Chromatin, 2009, 2, 1 CrossRef CAS PubMed.
- I.-C. Cheng, Y.-J. Chen, C.-W. Ku, Y.-W. Huang and C.-N. Yang, J. Chem. Inf. Model., 2015, 55, 2178–2186 CrossRef CAS PubMed.
- M. Abbasi, H. Sadeghi-Aliabadi, F. Hassanzadeh and M. Amanlou, J. Mol. Graphics Modell., 2015, 61, 186–195 CrossRef CAS PubMed.
- T. Agarwal, N. Annamalai, T. K. Maiti and H. Arsad, Gene, 2015, 580(1), 17–25 CrossRef PubMed.
- C. Estarellas, M. Otyepka, J. Koča, P. Banáš, M. Krepl and J. Šponer, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 1072–1090 CrossRef CAS PubMed.
- R. Sharma and G. N. Sastry, PLoS One, 2015, 10, e0144294 Search PubMed.
- N. Tuncbag, O. Keskin and A. Gursoy, Nucleic Acids Res., 2010, 38, W402–W406 CrossRef CAS PubMed.
- X. Zhu and J. C. Mitchell, Proteins, 2011, 79, 2671–2683 CrossRef CAS PubMed.
- J. Kenneth Morrow and S. Zhang, Curr. Pharm. Des., 2012, 18, 1255–1265 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- D. Case, T. Darden, T. Cheatham III, C. Simmerling, J. Wang, R. Duke, R. Luo, R. Walker, W. Zhang and K. Merz, AMBER 12, University of California, San Francisco, CA, 2012 Search PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 3, 198–210 CrossRef.
- J. Tang, J. G. Park, C. B. Millard, J. J. Schmidt and Y.-P. Pang, PLoS One, 2007, 2, e761 Search PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- R. W. Hockney and J. W. Eastwood, Computer simulation using particles, CRC Press, 1988 Search PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- W. van Gunsteren and H. Berendsen, Mol. Phys., 1977, 34, 1311–1327 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Equations to estimate RMSD, RMSF, and PCA; missing segments in the initial structures of WT and Y57A mutant; reproduced and aligned PDB structures for comparison; hydrogen bond occupancy along the long helix of Sgf11. See DOI: 10.1039/c6ra12647b |
| This journal is © The Royal Society of Chemistry 2016 |