
Role of LiTFSI in high Tg triphenylamine-based hole transporting material in perovskite solar cell
Abstract
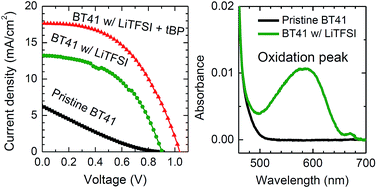
A hole transporting material based on triphenylamine with a high glass transition temperature (Tg) of 99 °C, coded as BT41, was synthesized and applied to a perovskite solar cell. The pristine BT41 showed a low power conversion efficiency (PCE) of 1.1% due to a low photocurrent density (Jsc) of ca. 6 mA cm−2 and an almost negligible fill factor of less than 0.2, which was significantly improved to 9.0%, however, owing mainly to the 3-fold improved Jsc of 17.6 mA cm−2, by adding both tert-butylpyridine (tBP) and lithium bis(trifluoromethane sulfonyl) imide (LiTFSI) as additives. The oxidation of BT41 was dominated by LiTFSI, which was responsible for the hole mobility increasing by one order of magnitude. The addition of the additive also reduced the recombination resistance, which correlates with the higher fill factor. Although both additives in BT41 contributed cooperatively to the improvement of the photovoltaic performance, LiTFSI played the major role in the enhancement.
 Please wait while we load your content...
Please wait while we load your content...

