DOI:
10.1039/C6RA12306F
(Paper)
RSC Adv., 2016,
6, 66180-66190
Fabrication of a novel bio-inspired collagen–polydopamine hydrogel and insights into the formation mechanism for biomedical applications†
Received
12th May 2016
, Accepted 1st July 2016
First published on 6th July 2016
Abstract
The bio-inspired approach to the construction of hydrogels with both excellent biological properties and superior initiative adhesive ability to cells is a crucial intersection of the branches of biomaterials science and biotechnology. In the present work, a novel bio-inspired collagen–polydopamine (COL–PDA) hydrogel has been successfully fabricated via collagen self-assembly and the incorporation of PDA. Systematic FTIR and XRD analysis confirmed that the hydrogen bond interactions between collagen and PDA did not destroy the triple helix conformation of collagen which is mainly responsible for the good biological properties of the COL–PDA hydrogel. In comparison with a pristine collagen hydrogel, the physicochemical properties (i.e. porosity, swelling ratio and water holding capacity) of COL–PDA hydrogels could be tuned by modulating the extent of interaction between COL and PDA with the change of dopamine concentrations. The further analysis of AFM observation indicated that a higher dopamine concentration could interrupt the aggregation or self-assembly of collagen molecules into fibrils via the extensive self-polymerization process of dopamine. Furthermore, the formation of a fibrous network could also be controlled by the self-assembly of collagen through varying the dopamine concentration, thus to adjust the thermal stability, enhance the resistance ability to enzymatic degradation and further cause promoted chain entanglement and forming increased elasticity. In addition, owing to the combined biological properties of COL and PDA, fabrication of a bio-inspired COL–PDA hydrogel has significant potential for the development of novel collagen hydrogels with good biological property and initiative adhesive ability to cells in biomedical applications.
Introduction
Collagen, the most abundant structural protein in the human body,1–3 has a long history as a natural material for biomedical applications.4–6 Due to its good biological properties7–9 (desirable biocompatibility, biodegradability and low inflammatory response), collagen molecules can form hydrogels, films or sponges which can be used as hemostatic pads, wound dressings, grafts and scaffolds for surgery and tissue engineering.10–14 Recently, collagen hydrogels have gained great achievements as swelling controlled drug delivery systems, cartilage damage or skin injury restorations in clinical applications, which are attributed to collagen hydrogels' water-rich, porosity-high properties and the ability to maintain the spherical morphology of encapsulated cells. These characteristics are benefit for efficient absorption of nutrition and elimination of metabolite.15–19 Furthermore, in additional to collagen hydrogels' material properties, there are increasing appreciations of the biological importance of collagen hydrogels in cell signaling and regulation of extracellular matrix (ECM) function. And it has been well illustrated that the typical triple helix structure (contiguous (Gly-Xaa-Yaa)n sequences) within collagen hydrogels could interact with cell surface receptors i.e. integrins (α2β1, α1β1, α10β1, α11β1), discoidin domain receptors (DDR1 and DDR2), mannose receptors and osteoblast receptor, that could regulate many cellular process including adhesion, proliferation and migration.20,21 The combination of its natural hierarchical physical structures and biological interactions makes collagen hydrogel an attractive candidate for a wide range of biomedical applications. However, with the increasing requests for collagen hydrogels in biomedical applications, some suitable chemical or physical modifications are still necessary to enhance collagen hydrogels' biological property, especially the initiative cell adhesive ability in cell culture system.22–25 Many literatures have also reported that although cells could adhere onto the surface of collagen hydrogels via the passive signal recognition,5,21,26 the initiative adhesive ability of collagen hydrogels is still not enough to satisfy the initial cells' adhesive demand, thus leading to poor cells adhesion and interactions within collagen hydrogels.3,20,27,28 Hence, developing a novel hydrogel with both superior biological property like collagen and good initiative adhesive ability to cells could be of great interest in biomedical applications.
Recently, mussel adhesive proteins (MAPs) excreted by marine mussels have attracted considerable attentions for their ability to form strong adhesive interactions with a wide range of substrates in wet environment.29–31 This is mainly attributed to the catechol group of dopa and a few lysine residues in MAPs.32 Although MAPs possess the superior adhesive ability, the expensive extracting process may hinder its practical application.33–35 Interestingly, Lee Haeshin and his group creatively applied polydopamine (PDA) with both the catechol group of dopa and the lysine residues to mimic MAPs' superior adhesive ability, which can form covalent or non-covalent bonds (hydrogen bond, van der Waals force) with organic or inorganic materials.36,37 Subsequently, PDA coatings have attracted considerable interests for a variety of biomedical applications from drug delivery or biosensing to tissue engineering. And this approach has many advantages such as simple and fast deposition onto virtually any surfaces, the opportunity for postfunctionalization via thiols and amines, biocompatibility, and so forth.29,38,39 However, the present studies mainly focus on the fabrication of adhesive bio-inert materials, such as silica, titanium, copolymer poly(ethylene imine)-graft-poly(ethylene glycol).35,36 Besides, there are only a few reports which have shown that PDA was codeposited with another bioactive biopolymer to form a functional surface with both the superior adhesive ability of PDA and the good biological activity, for example, the natural biopolymer hyaluronic acid/chitosan.40 In terms of applications, these results show up the possibility to incorporate PDA with collagen (COL) to fabricate a novel COL–PDA hydrogel that possesses both good biological property and initiative adhesive ability.
In the present work, a novel bio-inspired collagen hydrogel has been successfully fabricated via collagen self-assembly and the incorporation of PDA. Also the physicochemical properties, i.e. swelling behavior, porosity, water holding capacity, thermal stability, biodegradability and dynamic rheological behavior of COL–PDA hydrogels were measured. Meanwhile, the interactions between collagen and PDA, especially the structure integrity of collagen triple helix and the self-assembly of collagen molecules in the presence of PDA, were firstly investigated via Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD) and atomic force microscopy (AFM). Furthermore, the biocompatibility and cells adhesive ability of COL–PDA hydrogels were determined by 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. This cost-effective, superior biocompatibility and good cell adhesive ability of COL–PDA hydrogels would make it a very promising collagenous scaffold for biomedical applications.
Methods and materials
Extraction of collagen
Collagen used in this work was extracted from the skin of fresh grass carp (Ctenopharyngodon idella) which was purchased from the local slaughter house, according to our previous method.41 Note that the extraction process was conducted under 4 °C. Briefly, thawed fish skins were cut into 1 cm × 1 cm fragments, immersed into 0.01 M NaOH solution for 8 h (the mass ratio of grass carp skins to solution was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) to remove non-collagenous proteins and the solution was changed every two hours, then they were washed thoroughly with distilled water until the pH value was neutral. The degrease process was performed by stirring the skins for 8 hours in 10% (by weight) isopropyl alcohol followed by washing in distilled water for collagen extraction. Firstly, the pretreatment fish skins were stirred in 0.5 M acetic acid containing pepsin (pepsin 1:3000 from Sigma, USA) for two days; the pepsin concentration was set to be 2 wt% (dried base) of the fish skins, then centrifuged at 10000 rpm for 10 min. The supernatant was purified by salting-out with 1.5 M (NH4)2SO4 after adjusting pH to7.5, condensation by centrifugation at 10000 rpm for 10 min again and subsequent solubilized in 0.5 M acetic acid. Thus, the collagen solution obtained was dialyzed in dialysis tubes (Biosharp dialysis tubing, MWCO 12000–14000RC, diameter 36 mm, USA) against sterile 0.04 M disodium dihydrogen pyrophosphate acetic acid and distilled water for 2 days respectively. Then the collagen extracted was lyophilized and stored at room temperature. Further, the extracted collagen was analyzed by circular dichroism (model 400, AVIV) to verify the structural integrity of collagen as shown in Fig. 1. Dopamine hydrochloride and type I collagenase were purchased from Sigma-Aldrich. All other reagents used were analytical grade.
:20) to remove non-collagenous proteins and the solution was changed every two hours, then they were washed thoroughly with distilled water until the pH value was neutral. The degrease process was performed by stirring the skins for 8 hours in 10% (by weight) isopropyl alcohol followed by washing in distilled water for collagen extraction. Firstly, the pretreatment fish skins were stirred in 0.5 M acetic acid containing pepsin (pepsin 1:3000 from Sigma, USA) for two days; the pepsin concentration was set to be 2 wt% (dried base) of the fish skins, then centrifuged at 10000 rpm for 10 min. The supernatant was purified by salting-out with 1.5 M (NH4)2SO4 after adjusting pH to7.5, condensation by centrifugation at 10000 rpm for 10 min again and subsequent solubilized in 0.5 M acetic acid. Thus, the collagen solution obtained was dialyzed in dialysis tubes (Biosharp dialysis tubing, MWCO 12000–14000RC, diameter 36 mm, USA) against sterile 0.04 M disodium dihydrogen pyrophosphate acetic acid and distilled water for 2 days respectively. Then the collagen extracted was lyophilized and stored at room temperature. Further, the extracted collagen was analyzed by circular dichroism (model 400, AVIV) to verify the structural integrity of collagen as shown in Fig. 1. Dopamine hydrochloride and type I collagenase were purchased from Sigma-Aldrich. All other reagents used were analytical grade.
 |
| | Fig. 1 CD spectra of extracted fish collagen, the positive peak (maximum) at 221 nm and negative peak (minimum) at 202 nm indicating the structure integrity of pristine collagen. | |
Preparation of COL–PDA hydrogels
The acid soluble collagen solutions (10 mg ml−1) was freshly prepared and then the pH of collagen solution was adjusted to neutral (7.4 ± 0.2) by adding 2 M NaOH at 4 °C. Subsequently, dopamine was added to 2 ml of collagen solution in non-coated 24-well plates to obtain a final solution with a series of dopamine concentrations of 0.5 mg ml−1, 1 mg ml−1, 2 mg ml−1, 5 mg ml−1 and 10 mg ml−1. Then the above solutions were stirred at 4 °C for about 10 min. Finally, the mixture was immediately placed in an incubator at 37 °C for 4 h to initiate gelation of the collagen.
Porosity
The porosity of COL–PDA hydrogel was determined according to our previous work.41 Firstly, a 50 ml bottle was filled with ethanol and weighted as m1. Then, the lyophilized sample (m0) was immersed into the bottle which was later degassed by supersonic treatment to make the hydrogel infiltrated by ethanol and the whole weight was taken as m2. After that, the ethanol was removed from the surface of hydrogel gently and the weight of left part was measured as m3. The porosity of hydrogel was calculated by the following formula:| |
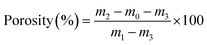 | (1) |
Swelling property and water holding capacity
The swelling capacity was measured at 25 °C by immersing the weighed (W0) lyophilized cylindrical samples in phosphate buffered saline (0.1 M PBS, pH7.4). After soaking for 1, 2, 3, 5, 8, 12, 20, 50, 90 min, the swollen hydrogels were taken out and immediately weighted (Wt) after removing the excess water. The swelling ratio (SR) can be calculated from the following formula:| |
 | (2) |
The water holding capacity (WHC) was examined according to the following method. In brief, the freshly prepared samples (equilibrium swollen hydrogel in water) were first weighted after wiping the water of the surface with filter paper slightly (Wswollen), then the samples were lyophilized, and weighted as Wlyophilized. The WHC can be calculated from the following formula:
| |
 | (3) |
Fourier tansform infrared spectroscopy (FTIR) analysis
The infrared spectrums of all lyophilized gel powders were obtained from tablets containing 2 mg of COL–PDA hydrogel sample in approximately 100 mg of potassium bromide (KBr) with an FTIR spectrophotometer (Nexus470, Nicolet, USA) in the range between 4000 and 400 cm−1 with a resolution of 4 cm−1.
X-ray diffraction (XRD) analysis
The X-ray diffraction pattern of the COL–PDA hydrogel was analyzed using CuKα radiation from a rotating anode generator operated at 40 kV and 40 mA in the range of 2θ (4–60°) with Mono-single filter with 10° min−1 scanning speed (D8 Advance, Bruker, Germany).
Thermal stability analysis
The thermal stability of the lyophilized COL–PDA hydrogel powders was assessed with DSC measurement (DSC 200 F3, Netzsch, Germany) over a temperature ranging from 20 to 180 °C. The samples (∼4 mg) were sealed in aluminum pans and heated at a maintained rate of 5 °C min−1 with an empty pan as the reference. The denaturation temperature (Td) was denoted as the endothermal peak temperature.
Enzymatic stability analysis
The in vitro enzymatic degradation of COL–PDA hydrogels was performed by exposing the freeze-dried specimens to bacterial type I collagenase according to the literature with slight modification.42 Firstly, each sample was incubated in 0.1 M Tris–HCl (pH 7.4) for 1 h at 37 °C and then 1 ml of 0.1 M Tris–HCl containing collagenase (200 U, Sigma) and 0.05 M CaCl2 was added to digest collagen for 24 h at 37 °C. The reaction was terminated by the addition of 0.2 ml of 0.25 M EDTA. The mixture was centrifuged for 5 min at 10000 rpm at 10 °C and the supernatant was collected. The hydroxyproline content was analyzed by ultraviolet spectroscopy. The biodegradation degree was determined as the percentage of the released hydroxyproline from the collagen to the completely degraded one without dopamine. The untreated collagen was used as a control.
Atomic force microscopy (AFM)
According to the previous work,43 the acid soluble collagen solutions (10 mg ml−1) were freshly prepared and then the pH of collagen solutions were adjusted to neutral (pH = 7.4 ± 0.2) by adding 2 M NaOH at 4 °C. Subsequently, dopamine was added to 2 ml of collagen solution in non-coated 24-well plates to obtain a final solution with a series of dopamine concentrations of 0.5 mg ml−1, 1 mg ml−1, 2 mg ml−1, 5 mg ml−1 and 10 mg ml−1. Then the above solutions were stirred at 4 °C for about 10 min. After diluting the original solutions 100 times (about 0.1 mg ml−1) and leaving it at 37 °C for 4 h, a droplet of the solution (about 0.5 μl) was deposited onto newly cleaved mica and then measured in tapping mode (Shimadzu SPM-9600, Japan).
Dynamic rheological measurements
The COL–PDA hydrogels (diameter φ = 20 mm, thickness = 5 mm) were prepared for dynamic viscoelasticity measurements. During the test, a rheometer (AR2000ex, TA, USA) with a parallel stainless steel plate (diameter = 40 mm, gap distance = 1 mm) was applied to obtain the values of the storage modulus (G′) and the loss modulus (G′′). Frequency sweeps measurements were carried out in a measuring mode with the frequency ranged from 0.01 Hz to 10 Hz at a constant strain of 2%, which was conducted within the range of linear viscoelastic region determined by strain sweeps measurements. The temperature was controlled by a Peltier temperature controller and a solvent trap was used to prevent the water loss of samples during the measurement. The value of tangentδ (tanδ) was also calculated as the G′′/G′ ratio, which reflected the thermal loss of energy.
Biocompatibility and cell adhesive ability analysis
Biocompatibility analysis
According to our previous work,41,44 L929 fibroblasts cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 100 units per ml penicillin, and 100 μg ml−1 streptomycin at 37 °C in humidified 95% air/5% CO2. The culture medium was changed every three days and the fibroblasts were used at passages three to eight. The mitochondria activity of the fibroblasts seeded on COL–PDA hydrogels was determined by colorimetric assay which detected the conversion of 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) to formazan. In brief, The fibroblasts were firstly seeded into the 24-well plates with COL–PDA hydrogels at a density of 1 × 104 cells per well, and cultured for 1, 3 and 5 days at 37 °C in an atmosphere of 5% CO2 while the cells cultured in saline and latex were used as the reference. At each time intervals, 50 μl per well of MTT solution (1 mg ml−1 in test medium) was added into the 24-well plates and incubated at 37 °C for 4 h to form the formazan crystals. After that, 400 μl per well of dimethylsulfoxide was added into the plates and mixed thoroughly to dissolve the dare blue crystals. Subsequently the solutions in 24-well plates were transferred into a 96-well plate, 150 μl per well. Finally, the 96-well plate was read on a microplate reader (Model550, Bio Rad Corp. U.S.A.) at a wavelength of 492 nm.
Cells adhesive ability of COL–PDA hydrogels
Firstly, L929 fibroblasts were seeded in the culture plates with COL–PDA hydrogels at the same concentration of 1 × 105 cells per well and cultured for 4 h, 8 h and 12 h. To remove un-adhesion fibroblasts, the cells were washed gently by PBS, then the cells were digested with trypsin (0.25 wt%) for approximate 2 min to obtain a cell suspension and counted using a hemacytometer. The attachment ratio of fibroblasts on protein coated culture plate was calculated using the following formula:
| Attachment ratio (%) = (number of cells adhesion on hydrogels/number of cells seeded on plates) × 100 |
Statistical analysis
The data were analyzed using the Statistical Analysis System Software V8 (SAS Institute Inc., Cary, NC, USA). Statistically significant differences were evaluated at p < 0.05 using Duncan's multiple range tests.
Results and discussion
Physicochemical properties of COL–PDA hydrogels
High porosity is usually considered significant for cell adhesion and proliferation in tissue engineering, which is benefit for cellular metabolism and supply of nutrients.11,12 Fig. 2A shows the change of the porosity of COL–PDA hydrogels with different dopamine concentrations (0.5 mg ml−1, 1 mg ml−1, 2 mg ml−1, 5 mg ml−1 and 10 mg ml−1). Among the hydrogels with different dopamine concentrations, the one in which COL–PDA hydrogel (dopamine concentration = 2.0 mg ml−1) exhibits the highest porosity (88.85% ± 0.95%), was much higher than that of pristine collagen hydrogel (27.48% ± 6.36%). It is worth noting that if we increased the dopamine concentration from 2 mg ml−1 to 10 mg ml−1, the porosity of COL–PDA hydrogel tends to decrease largely to 63.39% ± 3.21%. This irregular change of porosity along with the increasing of dopamine concentration may be ascribed to the oxidation polymerization of dopamine. It has been demonstrated that dopamine could polymerize to form PDA with chemical network structure under slightly alkaline condition,45,46 thus the formation of PDA could enhance the porosity of COL–PDA hydrogels at low dopamine concentration. However, since PDA could react with collagen via large amounts of hydrogen bonds, the porosity of COL–PDA hydrogels reduced at higher concentration of dopamine (5 mg ml−1 and 10 mg ml−1) suggests that the increasing amounts of PDA may retard the self-assembly of collagen, which leads to formation of heterogeneous pore structures with lower porosity. Fig. 2B illustrates the swelling ratio of COL–PDA hydrogel as a function of time (0–90 min). All the hydrogels show saturation after immersed in PBS solution within the first 20 min. And the swelling ratio increases slightly from 20 min to 50 min, and then almost keeps constant (80%–400%) from 50 min to 90 min, indicating that the swelling equilibrium reached. The incorporation of dopamine did promote the swelling ratio of COL–PDA hydrogels when the dopamine was lower than 2 mg ml−1, which is probably attributed to the formation of hydrogen bonds between the phenol hydroxyl groups of PDA and nitrogen atoms of collagen skeleton.47 However, the swelling ratio of COL–PDA with higher dopamine concentration (5 mg ml−1 and 10 mg ml−1) is lower than that of pristine collagen hydrogel. This may be ascribed to the extensive self-polymerization of dopamine that limits the extension of collagen fibrils to obtain a lower swelling ratio in PBS solution. Fig. 2A also shows the water-holding capacity of COL–PDA hydrogels. The water holding capacity of COL–PDA hydrogels increases slightly from 95.17 ± 0.09% of pristine collagen hydrogel to 95.58 ± 0.08% of COL–PDA hydrogel (dopamine concentration = 2 mg ml−1), then the water holding capacity decreases to 95.31 ± 0.21% and 94.30 ± 0.06% of COL–PDA hydrogels with dopamine concentration of 5 mg ml−1 and 10 mg ml−1, respectively. Note that, the water holding capacity of hydrogel is always considered important for cells' growth since higher water holding capacity means higher capturing ability of nutrients when the hydrogels are applied as tissue engineering scaffolds.28 However, no significant difference is found among the COL–PDA hydrogel groups, which indicates that the water holding capacity of COL–PDA hydrogels still maintains to a large extent after the incorporation of dopamine. Additionally, the morphology of pristine collagen hydrogel and COL–PDA hydrogels with dopamine concentrations of 0.5 mg ml−1, 1 mg ml−1, 2 mg ml−1, 5 mg ml−1 and 10 mg ml−1 were observed by digital camera (Fig. 2C). The colors of COL–PDA hydrogels change from white to black-brown along with the increase of dopamine concentration while the pristine collagen hydrogel is still white, which indicates the successful incorporation of dopamine to COL–PDA hydrogels.
 |
| | Fig. 2 (A) Porosity and water holding capacity of COL–PDA hydrogels (different capital letters indicate significant differences among different dopamine concentrations (p < 0.05)); (B) swelling ratio of COL–PDA hydrogels; (C) digital photo of pristine collagen hydrogel and COL–PDA hydrogels. | |
Influence of PDA on the structure integrity of collagen
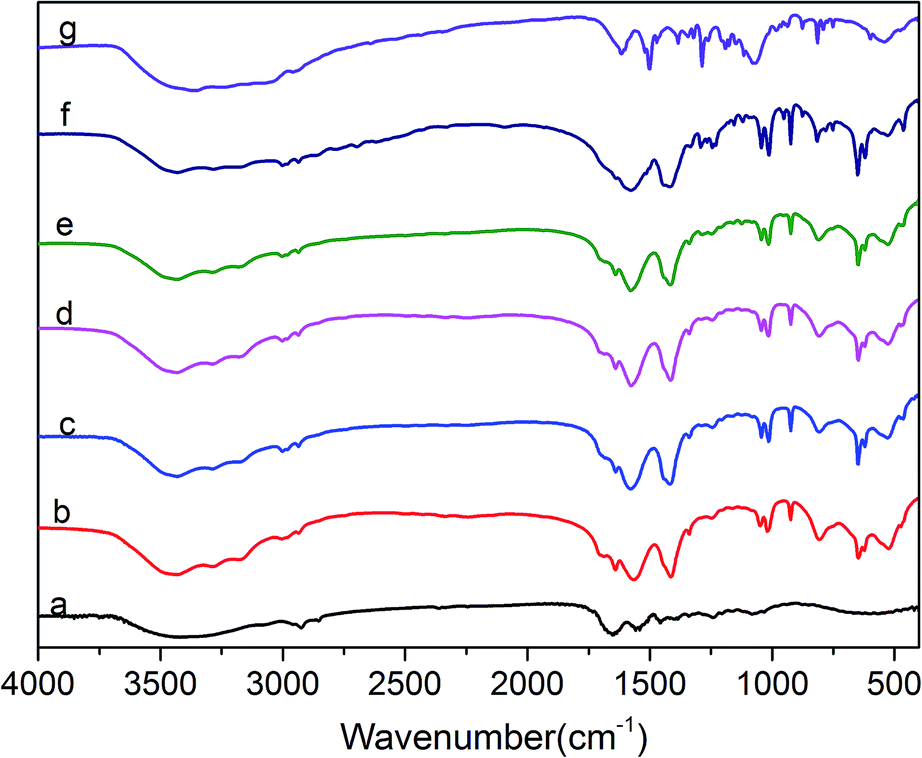
The FTIR spectra of protein molecules can be correlated directly to their backbone conformation, and it is commonly accepted that the secondary backbone conformation of collagen can be reflected in the FTIR spectra. Fig. 3 displays the FTIR spectra of COL–PDA hydrogels with different dopamine concentrations (0 mg ml−1, 0.5 mg ml−1, 1 mg ml−1, 2 mg ml−1, 5 mg ml−1, 10 mg ml−1), while pristine PDA and collagen are considered as the control. As revealed before, the intact collagen with typical triple helix conformation is characterized by its feature amide bonds in FTIR spectra. Normally, the amide A and B bonds at 3350 cm−1 and 3087 cm−1, respectively, are mainly ascribed to the stretching vibrations of N–H groups.24,41 The amide I bond at 1650 cm−1 is dominantly attributed to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, whereas the amide II absorption is due to amide N–H bending vibrations and C–N stretching vibrations. Meanwhile, the amide III peak is complex and assigned to the C–N stretching and N–H bending vibrations from amide linkages, as well as wagging vibrations of CH2 groups in the glycine backbone and proline side chains. Fig. 3 indicates that the positions of these major amide bonds do not change with the increasing dopamine concentration, particularly the amide I and II bands related to collagen triple helix maintain at the almost same position.41,44 Compared with the original collagen hydrogel, some new peaks appear in the COL–PDA hydrogels. The peaks at ∼1610 cm−1 and ∼1490 cm−1 are attributed to the overlap of the CC resonance vibration in the aromatic ring and the N–H bending vibration of PDA, respectively.47 Also the obvious peak at ∼1410 cm−1 ascribed to C–O–H of catechol groups of PDA, indicates that PDA has been successfully introduced into collagen hydrogel system. Furthermore, the peak at around 1350 cm−1 which appears as the shoulder of all COL–PDA hydrogels is assigned to indole ring (CNC) stretching modes. And the presence of indole features in the bulk PDA film supports the proposed structure of melanin-like polymer (PDA) consisting of 5,6-dihydroxyindole/5,6-indolequinone units, as is expected in the formation of PDA within COL–PDA hydrogels.45,48 Additionally, the region related to O–H and N–H stretching bonds, including typical amide bond A (∼3420 cm−1) and B (∼3093 cm−1) of collagen, appear to be broadened, which is mainly ascribed to the intermolecular hydrogen bonds formed between collagen and PDA with abundant –OH groups.49,50
O groups, whereas the amide II absorption is due to amide N–H bending vibrations and C–N stretching vibrations. Meanwhile, the amide III peak is complex and assigned to the C–N stretching and N–H bending vibrations from amide linkages, as well as wagging vibrations of CH2 groups in the glycine backbone and proline side chains. Fig. 3 indicates that the positions of these major amide bonds do not change with the increasing dopamine concentration, particularly the amide I and II bands related to collagen triple helix maintain at the almost same position.41,44 Compared with the original collagen hydrogel, some new peaks appear in the COL–PDA hydrogels. The peaks at ∼1610 cm−1 and ∼1490 cm−1 are attributed to the overlap of the CC resonance vibration in the aromatic ring and the N–H bending vibration of PDA, respectively.47 Also the obvious peak at ∼1410 cm−1 ascribed to C–O–H of catechol groups of PDA, indicates that PDA has been successfully introduced into collagen hydrogel system. Furthermore, the peak at around 1350 cm−1 which appears as the shoulder of all COL–PDA hydrogels is assigned to indole ring (CNC) stretching modes. And the presence of indole features in the bulk PDA film supports the proposed structure of melanin-like polymer (PDA) consisting of 5,6-dihydroxyindole/5,6-indolequinone units, as is expected in the formation of PDA within COL–PDA hydrogels.45,48 Additionally, the region related to O–H and N–H stretching bonds, including typical amide bond A (∼3420 cm−1) and B (∼3093 cm−1) of collagen, appear to be broadened, which is mainly ascribed to the intermolecular hydrogen bonds formed between collagen and PDA with abundant –OH groups.49,50
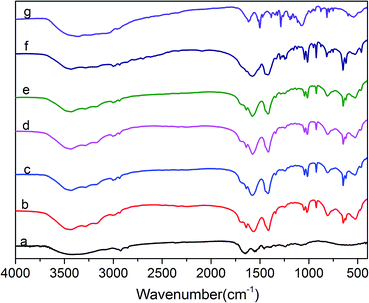 |
| | Fig. 3 FTIR spectra of COL–PDA hydrogels with different concentrations of dopamine ((a) 0 mg ml−1; (b) 0.5 mg ml−1; (c) 1 mg ml−1; (d) 2 mg ml−1, (e) 5 mg ml−1; (f) 10 mg ml−1 and (g) pristine PDA). | |
XRD spectrums were obtained and used to further investigate the structure integrity of collagen after incorporation of dopamine. Fig. 4 displays diffraction series corresponding to axial repeating structures. For collagen with structure integrity, the first position of the strong reflection at approximately 0.29 nm (2θ = 33°) relates to the axial rise distance (helical rise per residue) between the amino acid residues along collagen molecular triple helices, which is always considered crucial to the conformational integrity of collagen.51 The second broad peak at around 20° is associated with the diffuse scattering, while the third peak at approximately 1.2 nm (2θ = 5°) indicates an intermolecular lateral packing distance between the collagen molecular chains.41,52 Fig. 4 shows that the helical rise per residue value of the collagen molecules appears to be relatively unchanged following different dopamine concentrations, indicating the structural integrity of collagen within COL–PDA hydrogels still maintained to a large extent. However, slight changes can still be found that the typical diffuse scattering patterns at around 20° of COL–PDA hydrogels with 5 mg ml−1 and 10 mg ml−1 dopamine concentration appear to be more broadened than that of pure collagen hydrogel, which may be mainly caused by an increase in the disorder, or a reduction in the crystallite size, since the self-assembly of collagen during the fabrication of hydrogels could be partially disrupted by incorporation of dopamine via a large amount of hydrogen bonds forming between collagen side chains and PDA.53
 |
| | Fig. 4 X-ray diffraction (XRD) spectrum of COL–PDA hydrogels with different concentrations of dopamine ((a) 0 mg ml−1; (b) 0.5 mg ml−1; (c) 1 mg ml−1; (d) 2 mg ml−1, (e) 5 mg ml−1 and (f) 10 mg ml−1). | |
Enzymatic degradation property of COL–PDA hydrogels
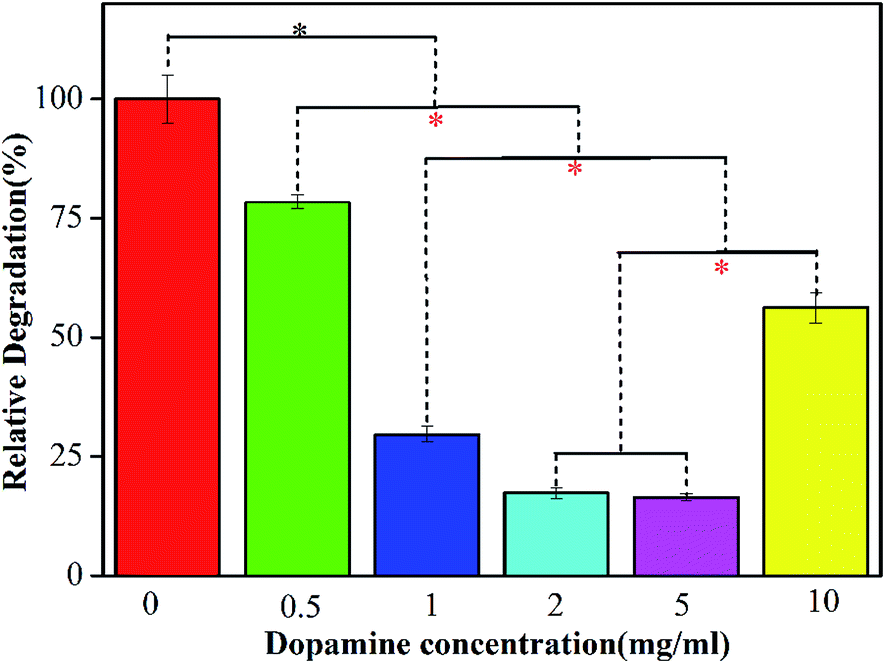
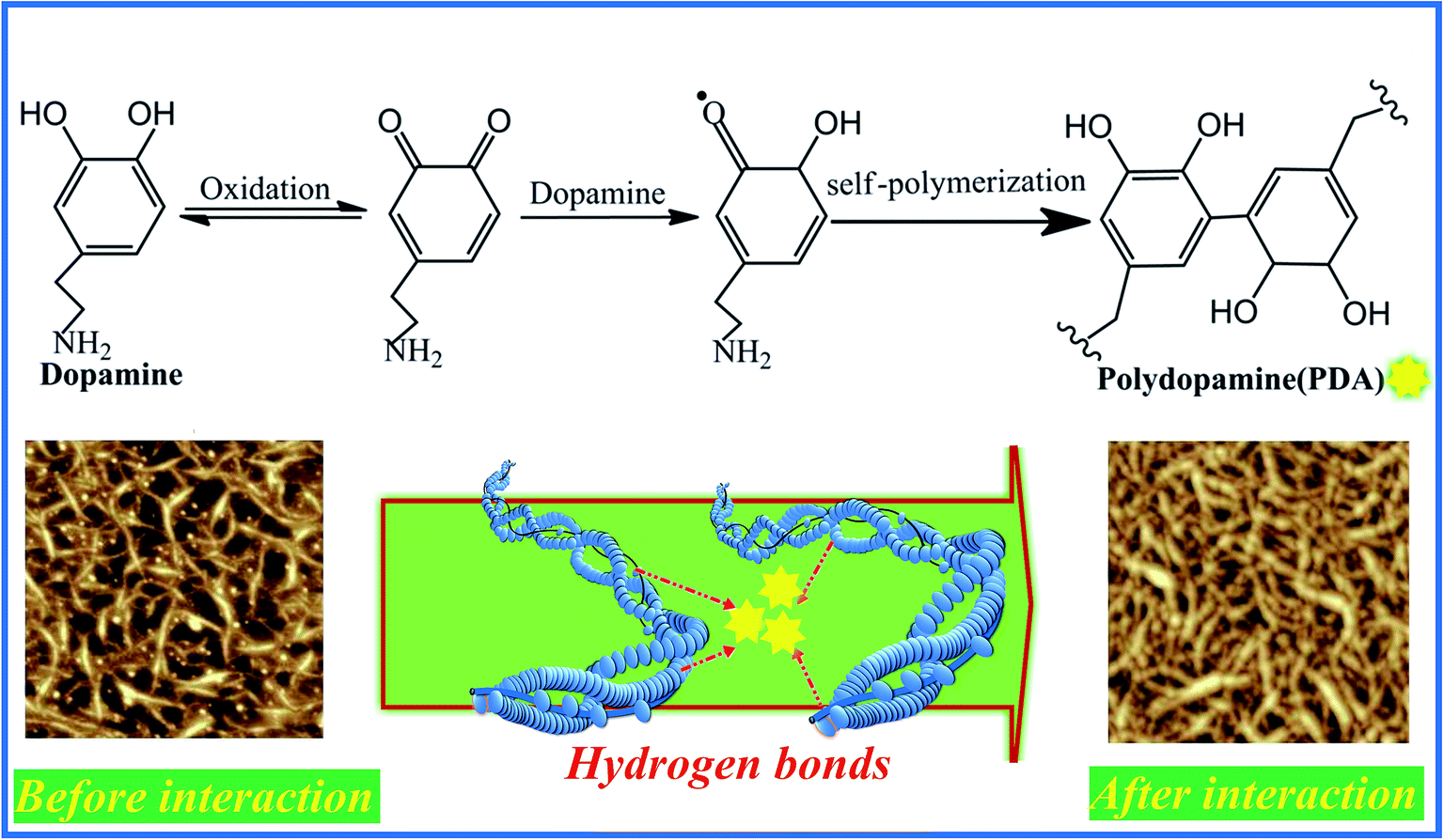
Collagen hydrogel, as one of the promising natural biopolymers with inter-connected three dimensional hierarchical structure, has drawn many of researchers' attentions in tissue engineering application, owing to its superior biocompatibility, low antigenicity and good bioactivity.4,20,41,44 One of our present work aims to firstly evaluate the effect of incorporation of dopamine on the enzymatic degradation property of COL–PDA hydrogels. Fig. 5 illustrates the resistance ability to enzymatic degradation of COL–PDA hydrogels after the degradation of bacterial type I collagenase for 24 h. As is well known, collagen with integrity structure, i.e. the unique triple helical molecule consisting of two identical α1(I) chains and a different α2(I) chain, is hardly to be degraded by traditional protein enzyme in gentile solution condition. Therefore, it is more convincing to determine the enzymatic degradation property of COL–PDA hydrogels using collagenase. Compared with pristine collagen hydrogel, the COL–PDA hydrogels exhibit slower degradation rate than that of pristine collagen hydrogel (*p < 0.05), with about 21.5%, 70.3%, 82.6%, 83.5% and 43.8% remaining for dopamine concentrations of 0.5 mg ml−1, 1 mg ml−1, 2 mg ml−1, 5 mg ml−1 and 10 mg ml−1, respectively. As illustrated in the schematic image (Fig. 6), solubilized collagen molecules can self-assemble into fibrous structure in vitro with the characteristic axial periodic structure, i.e. each triple helix is staggered from its molecular neighbor by a multiple of 67 nm in the direction of the helix, and laterally, the helices are arranged quasi-hexagonally with respect to each other within the fibril.54 During the fabrication process of COL–PDA hydrogel, dopamine is introduced into the system when collagen solution is ready for self-assembly. The reason why lower dopamine concentration (less than 5 mg ml−1) could promote the enzymatic degradation property of COL–PDA hydrogels, while higher dopamine concentration (10 mg ml−1) not, which may be that PDA could react with collagen and collagen fibrils via plenty of hydrogen bonds (Fig. 6), thus to enhance the stability of COL–PDA hydrogels. However, higher dopamine concentration may partially disrupt the self-assembly of collagen to form more stable collagen fibrils.46,55,56 Hence, dopamine concentration of 2 mg ml−1 is more appropriate for COL–PDA fabrication based on the measurement of enzymatic degradation property.
 |
| | Fig. 5 Degradation property of COL–PDA hydrogels with different concentrations of dopamine ((a) 0 mg ml−1, (b) 0.5 mg ml−1, (c) 1 mg ml−1, (d) 2 mg ml−1, (e) 5 mg ml−1 and (f) 10 mg ml−1, respectively, * means a statistically significant difference compared with each other). | |
 |
| | Fig. 6 Schematic diagram showing the possible interaction mechanism of collagen and PDA. | |
Influence of PDA on collagen self-assembly
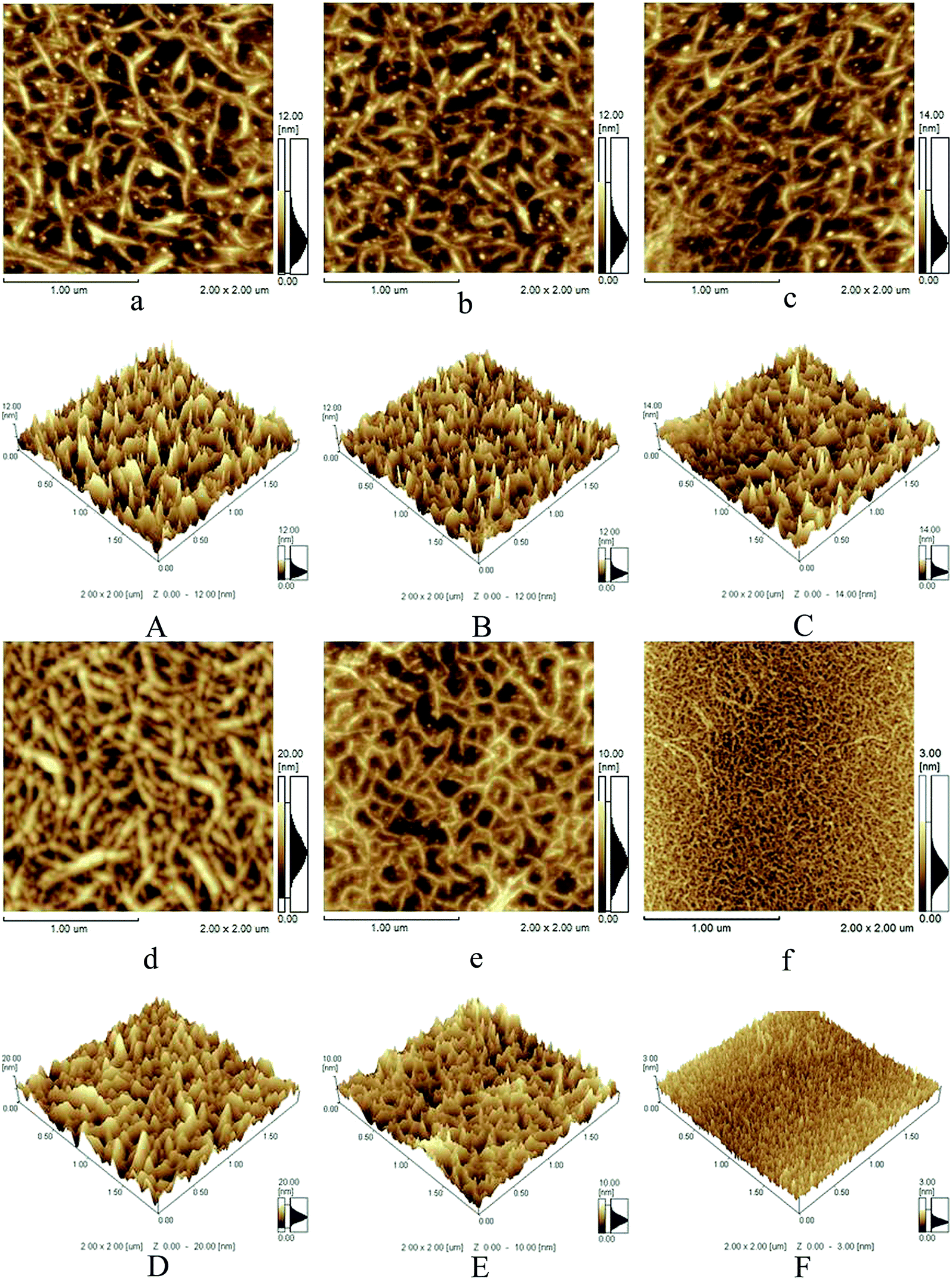
To further investigate the interaction between collagen and PDA, AFM has been used to analyze the collagen fibril behavior in the presence of PDA with different concentrations (0, 0.5, 1, 2, 5 and 10 mg ml−1). Firstly, the pristine collagen exhibits a typical fibrillar structure on the mica substrate with a homogenous and porous morphology of three-dimensional network, and many curved molecules or microfibrils are observed overlapping with one another, which is similar to our previous reports.43 Secondly, in the case of 0.5 and 1.0 mg ml−1 PDA, they appear to be relatively inhomogeneous and become slightly tight, and for 2 mg ml−1 PDA group, entangled fibrous network topographies are observed for the first time, indicating the occurrence of aggregation. Thirdly, for 5 and 10 mg ml−1 PDA groups, the density of the fibrils decreases and there are lots of fine fibrils compared with 1 and 2 mg ml−1 groups (Fig. 7c and d), especially for 10 mg ml−1 PDA group, bulky collagen fibrils are hardly observed (Fig. 7f), which suggests the assembly of collagen molecules is hindered seriously. The AFM images directly reveal the effect of PDA on collagen structure at the microfibril level. As reported by some literatures, many factors could affect the AFM morphology of collagen molecules, such as the drying mechanism, the interactions of van der Waals, hydrogen bond, ionic bond as well as covalent bond between collagen molecules and exogenous additions.41,57,58 In the present work, it is speculated that lower dopamine could take part in collagen fibril formation via large amounts of hydrogen bonds between the phenol hydroxyl groups of the self-polymerization (PDA) and nitrogen atoms of collagen skeleton as is reflected in the FTIR spectra, thus the collagen fibrous network with higher density and thicker diameter is observed (Fig. 7d). However, along with the increasing of dopamine concentration (5 or 10 mg ml−1), the extensive self-polymerization process of dopamine could disrupt the aggregation or assembly of collagen molecules into fibrils, further to induce the AFM topography revealing more fine and loose fibrils (Fig. 7e and f).
 |
| | Fig. 7 AFM phase images of COL–PDA hydrogels ((a) 0 mg ml−1, (b) 0.5 mg ml−1, (c) 1 mg ml−1, (d) 2 mg ml−1, (e) 5 mg ml−1 and (f) 10 mg ml−1, respectively) and AFM 3D images of COL–PDA hydrogels ((A) 0 mg ml−1, (B) 0.5 mg ml−1, (C) 1 mg ml−1, (D) 2 mg ml−1, (E) 5 mg ml−1 and (F) 10 mg ml−1, respectively). | |
Thermal properties of COL–PDA hydrogels
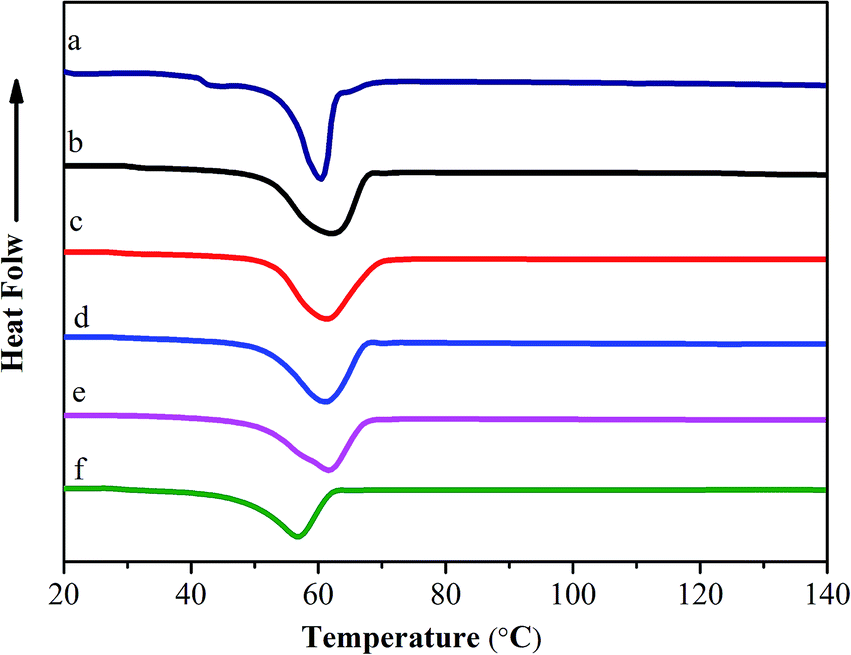
To further investigate the interaction between collagen and dopamine, differential scanning calorimetry (DSC) is used to detect the thermal stability of COL–PDA hydrogels, as shown in Fig. 8. Normally, the heat transformation of collagen is considered as the collapse of triple-helical structure of collagen into random coils, and the main endothermic peak is always assigned to Td.24,41 A typical endothermal peak from ∼40 °C to ∼80 °C presents the process of transformation of the skeleton structure of COL–PDA hydrogels. It is noted that if the structure integrity of collagen is destroyed, the endothermic peak shall transfer to lower temperature, thus the Td will be much lower than that of collagen with structure integrity.20,59 Fig. 8 shows that the Td of COL–PDA hydrogels keep almost the same endothermic peak as that of pristine collagen hydrogel (61.42 °C, 61.69 °C, 61.94 °C, 61.13 °C, 57.11 °C for COL–PDA hydrogels with 0.5 mg ml−1, 1 mg ml−1, 2 mg ml−1, 5 mg ml−1, 10 mg ml−1 dopamine concentration respectively and 60.32 °C for pristine collagen hydrogel). This phenomenon suggests that the typical triple helix of COL–PDA hydrogels is still preserved to a large extent. Additionally, larger endothermic peak areas of COL–PDA hydrogels are found than that of pristine collagen hydrogel, which means more energy is necessary to disrupt the skeleton structure of COL–PDA hydrogels during the DSC test process (Table S1†). However, higher dopamine concentration may prevent the self-assembling process and the alignment of collagen sequence to the three-dimensional packing structure as demonstrated by AFM images,54,60 further to decrease the Td of COL–PDA hydrogels.
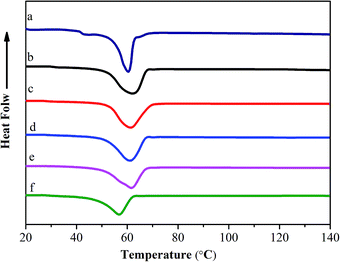 |
| | Fig. 8 DSC patterns of COL–PDA hydrogels with different concentrations of dopamine ((a) 0 mg ml−1; (b) 0.5 mg ml−1; (c) 1 mg ml−1; (d) 2 mg ml−1, (e) 5 mg ml−1 and (f) 10 mg ml−1). | |
Dynamic rheological properties of COL–PDA hydrogels
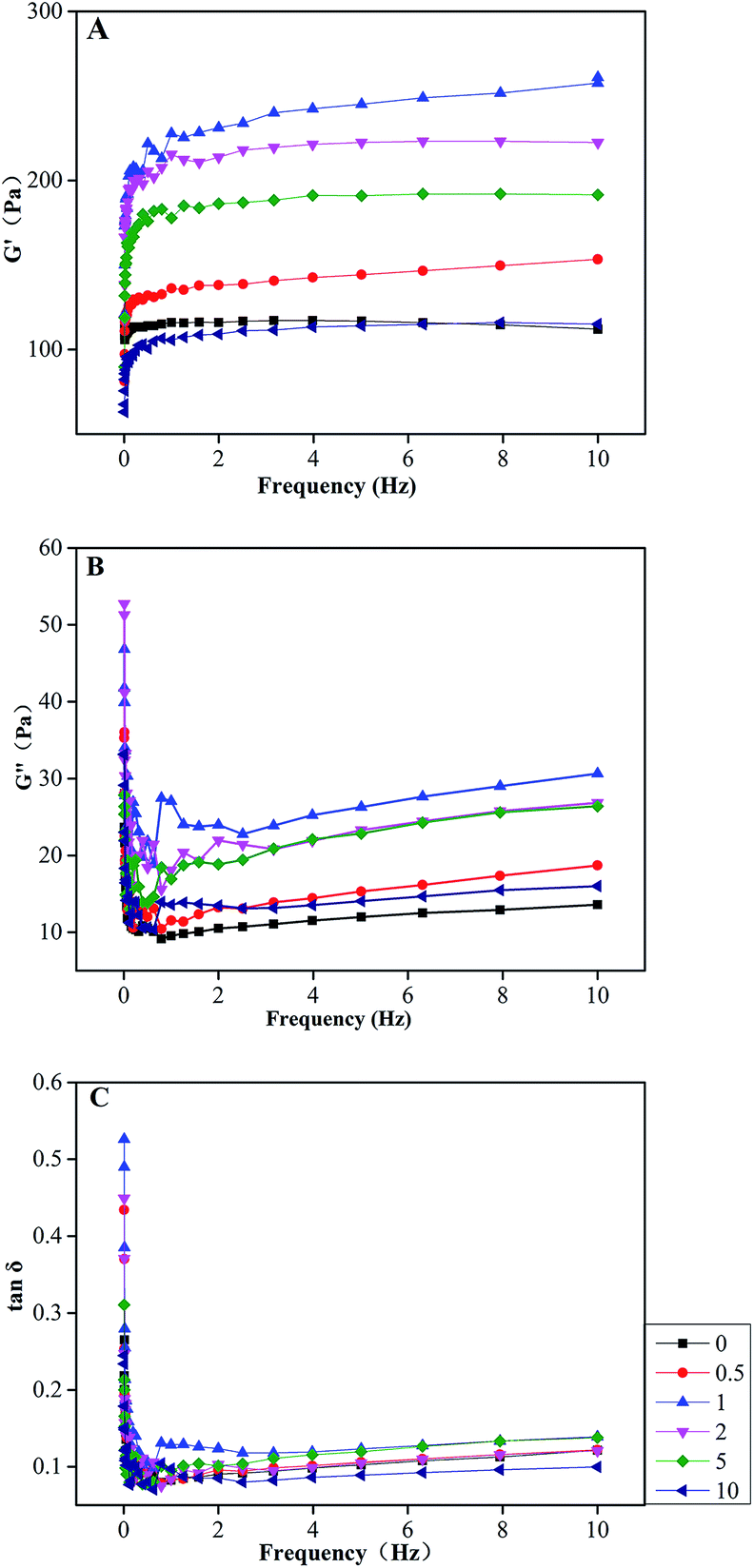
The storage (G′) and loss (G′′) moduli of a viscoelastic material represent the energy stored in and dissipated by the material, respectively. As using oscillatory rheometry exposing the solutions to a sinusoidal fluctuating strain, information about the structural properties of materials such as a solid and a liquid-like response can be obtained.61 The frequency-dependence curves of G′ and G′′ moduli of all COL–PDA hydrogels are demonstrated in Fig. 9A and B, respectively. As a function of dopamine concentration, both G′ and G′′ values of COL–PDA hydrogels increase to a maximum at 1.0 mg ml−1 dopamine before decreasing to a minimum at the highest dopamine concentration employed, 10 mg ml−1, whereas G′ was significantly greater than G′′ at the constant frequency, (i.e. G′ ≈ 277 Pa, whereas G′′ ≈ 27 Pa, the former is more ten times than latter at 1 Hz for hydrogels for 1 mg ml−1). The loss tangent (tanδ) is defined as the ratio G′′/G′, crossed the threshold (tanδ = 1) from liquid-like to solid-like behavior. Generally, the smaller the value of tanδ, the more apparent elastomeric behaviour performs.62 Note that, the values of tanδ of all samples are less than 1 (Fig. 9C) and exhibit a weak dependence of frequency, which indicate COL–PDA hydrogels show more stable gel-like properties. In addition, tanδ also demonstrates a similar tendency (increase to maximum at 1 mg ml−1 and then decrease to minimum at 10 mg ml−1) as a function of dopamine concentration. According to molecular chain entanglement theory,63 displacement of macromolecules such as collagen in high-concentration system is actually achieved by movement of polypeptide chains, during which it is inevitable the entanglement behavior occurs among the polypeptide chains, tangled parts known as entanglement points, contributing to formation of network structure showing a certain rubbery degree. When lower amounts of dopamine are introduced into the system, hydrogen bonds interaction between catechol groups of PDA and amino groups of collagen molecules may promote the combination among collagen monomers to some extent, generating more entanglement points, which can be demonstrated by AFM observations and degradation tests. However, after higher addition of dopamine, PDA formed by polymerization of dopamine blocked longitudinal alignment of collagen molecules and architecture of collagen fibrils, resulting in reduced chain entanglement and forming less entanglement points and decreased elasticity.
 |
| | Fig. 9 The storage moduli (A) and loss moduli (B) and loss tangent (C) of COL–PDA hydrogels as function of frequency. | |
Biocompatibility and cells adhesive ability of COL–PDA hydrogels
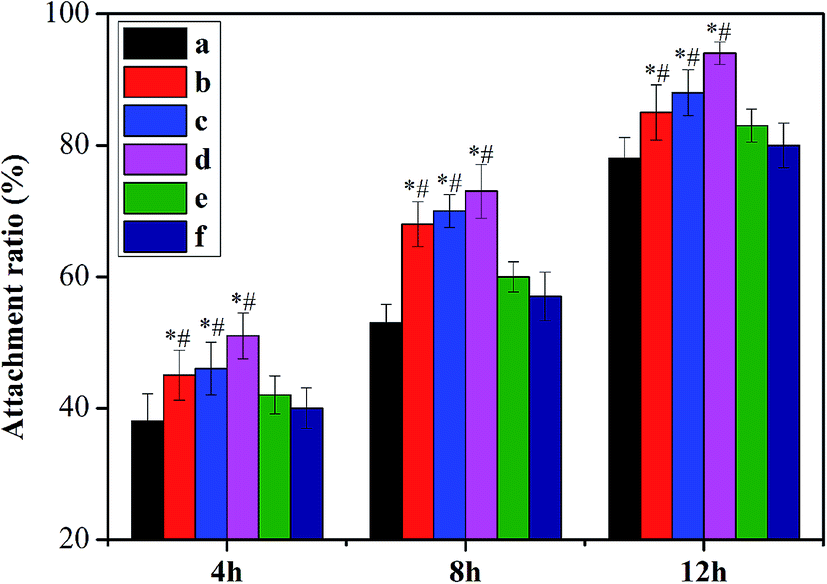
A potential tissue engineering scaffold provides the initial structural support and the template for cell adhesion and proliferation. Collagen has been widely investigated as a matrix, owing to its abundance in the ECM and the good biological property, i.e. good biocompatibility and low antigenicity. Although it is chemotactic to cells and in favor of cellular attachment, it is still not enough that collagen just adheres cells via weak affinity, and cell adhesion is always considered crucial of tissue engineering scaffold in the initial treatment. Hence, we have fabricated a novel bio-inspired collagen hydrogel with both good biocompatibility of collagen and good adhesive property of PDA in this work. Here, the biocompatibility of COL–PDA hydrogels is determined by the MTT assay. As shown in Fig. 10, the proliferation of L929 fibroblasts cultured on scaffolds increased in all experimental groups during the culture period compared with saline group (control), which indicates that both collagen itself and its modification (COL–PDA hydrogels) could promote growth and proliferation of cells. And the COL–PDA hydrogels with lower dopamine concentration (0.5 mg ml−1 to 2 mg ml−1) show higher proliferation rate than pristine collagen hydrogel, which may be induced by the higher adhesive ability (Fig. 11). However, COL–PDA hydrogels for higher dopamine concentration (5 mg ml−1 and 10 mg ml−1) reveal a relatively decreased cell proliferation rate that may be due to the fact that dopamine could induce the expression of antioxidative enzyme, thus to adjust the proliferation of cell in the cultural medium.64 Further, we evaluate the cell adhesion of both pristine collagen hydrogel and COL–PDA hydrogels in the initial 4 h, 8 h and 12 h culture periods. On one hand, the COL–PDA hydrogels with the dopamine concentration of 0.5 mg ml−1, 1 mg ml−1 and 2 mg ml−1 did promote the cell attachment ratio from 38 ± 4.2% to 45 ± 3.8%, 46 ± 4.0% and 51 ± 3.5%, respectively at 4 h. This may mainly depend on the good adhesive ability of PDA within COL–PDA hydrogel which has been demonstrated by other researchers.37 Whereas, higher dopamine concentration groups (5 mg ml−1 and 10 mg ml−1) unfortunately decrease the adhesive ability of COL–PDA hydrogels that may due to the fact that higher concentration of dopamine could inhibit cellular activity to some extent, thus further to decrease the cell adhesive ability of COL–PDA hydrogels. These results suggest that PDA with suitable concentration can work as an on-demand robust glue for creating a novel scaffold processing both the biocompatibility of collagen and the adhesive ability of PDA.
 |
| | Fig. 10 Cell proliferation on COL–PDA hydrogels ((a) 0 mg ml−1; (b) 0.5 mg ml−1; (c) 1 mg ml−1; (d) 2 mg ml−1, (e) 5 mg ml−1 and (f) 10 mg ml−1), saline group (control), and latex (positive control). The absorbance is normalized with that of saline group (control) at each time interval. *p < 0.05 compared with saline group (control). | |
 |
| | Fig. 11 Cell adhesion on COL–PDA hydrogels ((a) 0 mg ml−1; (b) 0.5 mg ml−1; (c) 1 mg ml−1; (d) 2 mg ml−1, (e) 5 mg ml−1 and (f) 10 mg ml−1) at 4 h, 8 h and 12 h. *p < 0.05 relative to the 0 mg ml−1 group (group a) and #p < 0.05 relative to the 5 mg ml−1 and 10 mg ml−1 group (group e and f). | |
Conclusions
Herein, we had explored a novel mussel inspired collagen hydrogel (COL–PDA hydrogel) via collagen self-assembly and incorporation of PDA, which possessed good biocompatibility and excellent cells adhesive ability when the dopamine concentration ranged from 0.5 mg ml−1 to 2 mg ml−1. The good biocompatibility of COL–PDA hydrogels was ascribed to the structure integrity of typical triple helix of collagen. And large amounts of hydrogen bonds between the phenol hydroxyl (C–OH) of PDA and nitrogen atoms (N–CO) of collagen skeleton took a great role in the interactions between PDA and collagen within COL–PDA hydrogels, which had been demonstrated by FTIR and XRD spectra. The incorporation of PDA was mainly responsible for the excellent cells adhesive ability of COL–PDA hydrogels. AFM observations revealed that higher dopamine concentration (5 mg ml−1 or 10 mg ml−1) could interrupt the aggregation or self-assembly of collagen molecules into fibrils via extensive self-polymerization process of dopamine, whereas suitable dopamine concentration could induce the formation of collagen fibrous network with higher density and thicker diameter, thus to increase the thermal stability, resistance ability to enzymatic degradation and further promote chains entanglement and form increased elasticity. Therefore, COL–PDA hydrogel with superior biocompatibility and good cell adhesive ability would endow it as a very promising collagenous scaffold for biomedical applications.
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 21506070; No. 51503129), the Fundamental Research Funds for the Central Universities (No. 2662015QC014; No. 2662014BQ053), Hubei Provincial Natural Science Foundation of China (No. 2015CFB391) and China Agriculture Research System (No. CARS-46-23).
Notes and references
- C. M. Murphy, M. G. Haugh and F. J. O'Brien, Biomaterials, 2010, 31, 461–466 CrossRef CAS PubMed.
- Y. Hu, L. Liu, Z. Gu, W. Dan, N. Dan and X. Yu, Carbohydr. Polym., 2014, 102, 324–332 CrossRef CAS PubMed.
- T. W. Hsiao, P. A. Tresco and V. Hlady, Biomaterials, 2015, 39, 124–130 CrossRef CAS PubMed.
- B. Walters and J. Stegemann, Acta Biomater., 2014, 10, 1488–1501 CrossRef CAS PubMed.
- B. An, Y. S. Lin and B. Brodsky, Adv. Drug Delivery Rev., 2016, 97, 69–84 CrossRef CAS PubMed.
- L. He and P. Theato, Eur. Polym. J., 2013, 49, 2986–2997 CrossRef CAS.
- B. Cuq, N. Gontard and S. Guilbert, Cereal Chem., 1998, 75, 1–9 CrossRef CAS.
- T. Muthukumar, D. Prakash, K. Anbarasu, B. S. Kumar and T. P. Sastry, RSC Adv., 2014, 4, 64267–64276 RSC.
- E. I. Alarcon, K. I. Udekwu, C. W. Noel, L. B. Gagnon, P. K. Taylor, B. Vulesevic, M. J. Simpson, S. Gkotzis, M. M. Islam, C. J. Lee, A. Richter-Dahlfors, T. F. Mah, E. J. Suuronen, J. C. Scaiano and M. Griffith, Nanoscale, 2015, 7, 18789–18798 RSC.
- H. Tian, Y. Chen, C. Ding and G. Li, Carbohydr. Polym., 2012, 89, 542–550 CrossRef CAS PubMed.
- M. Rutgers, D. B. Saris, L. A. Vonk, M. H. van Rijen, V. Akrum, D. Langeveld, A. van Boxtel, W. J. Dhert and L. B. Creemers, Tissue Eng., Part A, 2013, 19, 59–65 CrossRef CAS PubMed.
- L. Wang and J. P. Stegemann, Acta Biomater., 2011, 7, 2410–2417 CrossRef CAS PubMed.
- L. Cen, W. Liu, L. Cui, W. Zhang and Y. Cao, Pediatr. Res., 2008, 63, 492–496 CrossRef CAS PubMed.
- B. Hoyer, A. Bernhardt, A. Lode, S. Heinemann, J. Sewing, M. Klinger, H. Notbohm and M. Gelinsky, Acta Biomater., 2014, 10, 883–892 CrossRef CAS PubMed.
- M. Hamidi, K. Rostamizadeh and M. A. Shahbazi, in Intelligent Nanomaterials, John Wiley & Sons, Inc, 2012, pp. 583–624, DOI:10.1002/9781118311974.ch15.
- M. R. Kim and T. G. Park, J. Controlled Release, 2002, 80, 69–77 CrossRef CAS PubMed.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed.
- K. Madhavan, D. Belchenko and W. Tan, J. Biomed. Mater. Res., Part A, 2011, 97, 16–26 CrossRef PubMed.
- L. Zhang, K. Li, W. Xiao, L. Zheng, Y. Xiao, H. Fan and X. Zhang, Carbohydr. Polym., 2011, 84, 118–125 CrossRef CAS.
- X. Yu, C. Tang, S. Xiong, Q. Yuan, Z. Gu, Z. Li and Y. Hu, Curr. Org. Chem., 2016, 20, 1797–1812 CrossRef CAS.
- B. An and B. Brodsky, Blood, 2016, 127, 521–522 CrossRef PubMed.
- H. Liu, D. Li and S. Guo, Food Chem., 2007, 101, 621–625 CrossRef CAS.
- L. M. Delgado, Y. Bayon, A. Pandit and D. I. Zeugolis, Tissue Eng., Part B, 2015, 21, 298–313 CrossRef CAS PubMed.
- M. Zhang, J. Li, C. Ding, W. Liu and G. Li, Food Hydrocolloids, 2013, 30, 504–511 CrossRef CAS.
- C. Xu and Y. Wang, J. Adhes. Dent., 2012, 14, 11–18 CAS.
- Z. Bagher, M. Azami, S. Ebrahimi-Barough, H. Mirzadeh, A. Solouk, M. Soleimani, J. Ai, M. R. Nourani and M. T. Joghataei, Mol. Neurobiol., 2016, 53, 2397–2408 CrossRef CAS PubMed.
- X. Zhang, Y. H. Yang, J. R. Yao, Z. Z. Shao and X. Chen, ACS Sustainable Chem. Eng., 2014, 2, 1318–1324 CrossRef CAS.
- H. Lin, W. Dan and N. Dan, J. Appl. Polym. Sci., 2012, 123, 2753–2761 CrossRef CAS.
- S. M. Kang, S. Park, D. Kim, S. Y. Park, R. S. Ruoff and H. Lee, Adv. Funct. Mater., 2011, 21, 108–112 CrossRef CAS.
- S. Hong, K. Y. Kim, H. J. Wook, S. Y. Park, K. D. Lee, D. Y. Lee and H. Lee, Nanomedicine, 2011, 6, 793–801 CrossRef CAS PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- N. G. Rim, S. J. Kim, Y. M. Shin, I. Jun, D. W. Lim, J. H. Park and H. Shin, Colloids Surf., B, 2012, 91, 189–197 CrossRef CAS PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- L. Liu, B. Shao and F. Yang, Sep. Purif. Technol., 2013, 118, 226–233 CrossRef CAS.
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS.
- M. Shin, H. K. Kim and H. Lee, Biotechnol. Prog., 2014, 30, 215–223 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- H. Sun, M. Ai, S. Zhu, X. Jia, Q. Cai and X. Yang, RSC Adv., 2015, 5, 95631–95642 RSC.
- N. G. Rim, S. J. Kim, Y. M. Shin, I. Jun, D. W. Lim, J. H. Park and H. Shin, Colloids Surf., B, 2012, 91, 189–197 CrossRef CAS PubMed.
- X. Zhong, Y. Song, P. Yang, Y. Wang, S. Jiang, X. Zhang and C. Li, PLoS One, 2016, 11, 121–125 Search PubMed.
- S. Zhu, Z. Gu, Y. Hu, W. Dan and S. Xiong, J. Appl. Polym. Sci., 2016 DOI:10.1002/app.43550.
- Y. Hu, L. Liu, W. Dan, N. Dan, Z. Gu and X. Yu, Int. J. Biol. Macromol., 2013, 55, 221–230 CrossRef CAS PubMed.
- D. Li, C. Mu, S. Cai and W. Lin, Ultrason. Sonochem., 2009, 16, 605–609 CrossRef CAS PubMed.
- Y. Hu, L. Liu, W. Dan, N. Dan and Z. Gu, J. Appl. Polym. Sci., 2013, 130, 2245–2256 CrossRef CAS.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- P. Zhou, F. Wu, T. Zhou, X. Cai, S. Zhang, X. Zhang, Q. Li, Y. Li, Y. Zheng, M. Wang, F. Lan, G. Pan, D. Pei and S. Wei, Biomaterials, 2016, 87, 1–17 CrossRef CAS PubMed.
- C. Cao, L. Tan, W. Liu, J. Ma and L. Li, J. Power Sources, 2014, 248, 224–229 CrossRef CAS.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- X. Liu, J. Cao, H. Li, J. Li, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 9384–9395 CrossRef CAS PubMed.
- C. A. Maxwell, T. J. Wess and C. J. Kennedy, Biomacromolecules, 2006, 7, 2321–2326 CrossRef CAS PubMed.
- Y. Hu, L. Liu and D. Weihua, J. Soc. Leather Technol. Chem., 2013, 97, 200–206 Search PubMed.
- C. Ding, M. Zhang, K. Wu and G. Li, Polymer, 2014, 55, 5751–5759 CrossRef CAS.
- S. Perumal, O. Antipova and J. P. Orgel, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2824–2829 CrossRef CAS PubMed.
- F. W. Kotch and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3028–3033 CrossRef CAS PubMed.
- M. J. Buehler, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12285–12290 CrossRef CAS PubMed.
- L. He, C. Mu, J. Shi, Q. Zhang, B. Shi and W. Lin, Int. J. Biol. Macromol., 2011, 48, 354–359 CrossRef CAS PubMed.
- C. C. Ding, M. Zhang and G. Y. Li, Carbohydr. Polym., 2015, 119, 194–201 CrossRef CAS PubMed.
- H. Tan, B. Wu, C. Li, C. Mu, H. Li and W. Lin, Carbohydr. Polym., 2015, 129, 17–24 CrossRef CAS PubMed.
- J. He, Y. Su, T. Huang, B. Jiang, F. Wu and Z. W. Gu, Colloids Surf., B, 2014, 116, 303–308 CrossRef CAS PubMed.
- S. Monika and F. Wolfgang, Eur. J. Pharm. Biopharm., 2001, 51, 259–265 CrossRef.
- M. Korhonen, L. Hellen, J. Hirvonen and J. Yliruusi, Int. J. Pharm., 2001, 221, 187–196 CrossRef CAS PubMed.
- T. T. Hsieh, C. Tiu, G. P. Simon and R. Y. Wu, J. Non-Newtonian Fluid Mech., 1999, 86, 15–35 CrossRef CAS.
- X. Wang, Z. Gu, B. Jiang, L. Li and X. Yu, Biomater. Sci., 2016, 4, 678–688 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12306f |
| ‡ Authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.