
PEGylated graphene oxide-based nanocomposite-grafted chitosan/polyvinyl alcohol nanofiber as an advanced antibacterial wound dressing†
Abstract
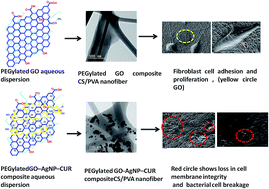
Designing composite nanomaterials that display multiple antibacterial mechanisms offers new prototype against bacterial resistance. This study presents a multi-component composite-based nanofiber embodying the antibacterial and physiochemical properties of silver nanoparticles (Ag NPs), graphene oxide (GO), chitosan (CS), and curcumin (CUR). Physiologically stable PEGylated GO–Ag NP–CUR nanocomposites were synthesized, with the PEGylated GO serving as the template. The as-synthesized nanocomposite was incorporated into the CS/polyvinyl alcohol (PVA) nanofiber. The successful formation and stability of the PEGylated-GO–Ag NP–CUR composite nanofiber were characterized by various techniques. The antibacterial potential of the PEGylated-GO–Ag NP–CUR composite nanofiber was evaluated and showed an enhanced antibacterial effect compared to various nanoformulations. The plausible antibacterial mechanism of the PEGylated-GO–Ag NP–CUR nanofiber was determined and depicted herein. The presence of GO in the composite nanofiber enhances its mechanical properties compared to CS/PVA nanofiber, with an ultimate tensile strength (UTS) of 25 MPa compared to 7.2 MPa and a Young's modulus (E) of 363.7 MPa compared to 73 MPa. The biocompatibility of the nanofiber mat was confirmed by in vitro cell viability assay. Therefore a facile approach for the design of a biocompatible wound dressing with enhanced mechanical and antibacterial property was explored and detailed herein.
 Please wait while we load your content...
Please wait while we load your content...

