
Methylene diphenyl diisocyanate (MDI) and toluene diisocyanate (TDI) based polyurethanes: thermal, shape-memory and mechanical behavior
Abstract
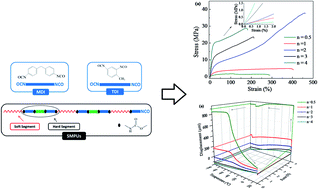
Shape memory polymers (SMPs) have attracted extensive attention from basic and fundamental research to industrial and practical applications. Among them, shape memory polyurethanes (SMPUs) have different applications such as in textile finishings, adhesives, coatings, automotive parts, furniture, construction materials, thermal insulation materials and footwear industries because they can be synthesized with different types of molecular architectures by manipulating their composition and properly choosing the chemical structure of their individual components. In this work, the synthesis and characterization of SMPUs, based on two-step polymerization, are reported. The hard segment of SMPU was composed of diisocyanate (toluene 2,4-diisocyanate (TDI) or 4,4′-methylene diphenyl diisocyanate (MDI)) and a chain extender, 1,4-butanediol (BD). On the other hand, the soft segment was prepared by a polyol, poly(oxytetramethylene) glycol (PTMG). By selectively choosing the hard-to-soft segment content, the glass transition temperature of SMPUs could be varied from −52.1 °C to 8.6 °C, while the proper combination of both segments imparts combined ductility and strength to our materials. Furthermore, the shape memory effect was found to depend on hydrogen bonding molecular interactions, making TDI-based SMPUs more appropriate for their commercial use.
 Please wait while we load your content...
Please wait while we load your content...

