
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of substituted γ- and δ-lactams based on titanocene(III)-catalysed radical cyclisations of trichloroacetamides†
Faïza
Diaba
a,
Enrique
Gómez-Bengoa
b,
Juan M.
Cuerva
c,
Josep
Bonjoch
*a and
José
Justicia
*c
aLaboratori de Química Orgànica, Facultat de Farmàcia, IBUB, Universitat de Barcelona, Av. Joan XXIII s/n, 08028-Barcelona, Spain. E-mail: josep.bonjoch@ub.edu
bDepartamento de Química Orgánica I, Universidad del País Vasco, Manuel Lardizábal 3, 20018 San Sebastián, Spain
cDepartamento de Química Orgánica, Facultad de Ciencias, Universidad de Granada, C. U. Fuentenueva s/n, 18071-Granada, Spain. E-mail: jjusti@ugr.es
First published on 1st June 2016
Abstract
A new procedure for the synthesis of γ- and δ-lactams based on a Cp2TiCl-catalysed cyclisation of trichloroacetamides under mild reaction conditions is reported. Theoretical studies supported the observed regioselectivity in the cyclisations and the mechanism involved in the dehalogenation process.
Since its characterization in 1972 by Green et al.,1 biscyclopentadienyltitanium(III) chloride, Cp2TiCl, has emerged as a useful tool in organic chemistry, being extensively applied in several synthetic processes. Seminal work by RajanBabu and Nugent, Gansäuer and others has shown that epoxides,2–4a allylic halides,4b and some carbonyl derivatives3d,e,4c,5 are suitable starting materials for monoelectronic reduction to the corresponding carbon-centered radicals. These radicals are involved in interesting processes,6 such as reduction reactions, addition to alkenes or alkynes, pinacol couplings, and Barbier-, Wurtz-, and Reformatsky-type reactions, which have also been applied in natural product synthesis.7 Recently, other functionalities have been included in the arsenal of Cp2TiCl chemistry. Thus, allylic and propargylic carboxylates,8 ozonides,9 nitriles,10 imines,11 chloroaminals,12 and hemiaminals13 are now used in relevant C–C bond-forming reactions. Within this context, the expansion of Cp2TiCl-based protocols to new, easy to handle functionalities would be highly desirable.
Our attention was recently attracted to trichloroacetamides, which have been used as dichloromethylcarbamoyl radical precursors to synthesize nitrogen-containing heterocycles14,15 and alkaloids,16 using either atom transfer radical cyclisations (ATRC) mediated by Cu(I),17 Ru(II),18 Ni–AcOH,19 and Fe(0)/FeCl3,20 or reductive methods based on Bu3SnH,21 and TTMSS.22 However, the use of titanium reagents to generate radical species from trichloroacetamides has not been reported so far.
With these antecedents in mind, we thought that Cp2TiCl would be able to generate the corresponding dichloromethylcarbamoyl radicals from trichloroacetamides under mild reaction conditions for their use in cyclisation reactions. Moreover, the final polyhalogenated compounds could be subsequently reduced by Cp2TiCl and a hydrogen atom source, directly yielding non-halogenated final products. It is worth noting that this transformation could be carried out using substoichiometric amounts of Cp2TiCl (Scheme 1).6
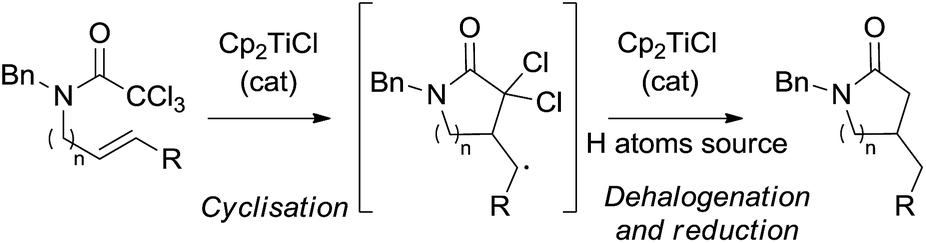 | ||
| Scheme 1 Proposed Cp2TiCl-catalysed radical cyclisation of trichloroacetamides. | ||
To check our hypothesis, we selected trichloroacetamide 1, which has been previously used in related syntheses of polyhalogenated lactams.14c,17c,d Compound 1 reacted in the presence of 3 equiv. of Cp2TiCl at room temperature to yield the corresponding γ-lactam 2, albeit in low yield (24%) (Scheme 2a), and minor amounts of monohalogenated derivative 3 (16%) were also isolated, both products arising from a 5-exo-trig cyclisation. The reduction of the intermediate radicals in this process could derive from the presence of adventitious water in the solvent (THF), via Ti(III)-aquacomplexes.23 This preliminary result indicated that our working hypothesis was correct. Nevertheless, three main drawbacks were observed: (i) the use of high amounts of Cp2TiCl, (ii) a mixture of reaction products, and (iii) a low yield. To overcome these disadvantages, we decided to study the cyclisation process using different substoichiometric amounts of Cp2TiCl and longer reaction times, in order to determine the influence of these factors on the yields of the final products. In these studies, we used the aprotic combination of Mn dust and Me3SiCl/2,4,6-collidine (Coll) as a regenerating agent of titanocene(III) species (see Scheme 2b, protocol A).4a,6 Additionally, it is worth noting that the reduction of the generated radicals required a hydrogen atom source. Thus, we also checked the cyclisation of 1 in the presence of an efficient hydrogen atom source such as H2O, using substoichiometric amounts of Cp2TiCl (Scheme 2b, protocol B). In this case, the regenerating agent of the titanocene(III) species was the mixture of Mn dust and 2,4,6-collidine hydrochloride (Coll·HCl).3a–c The results are depicted in Table 1.
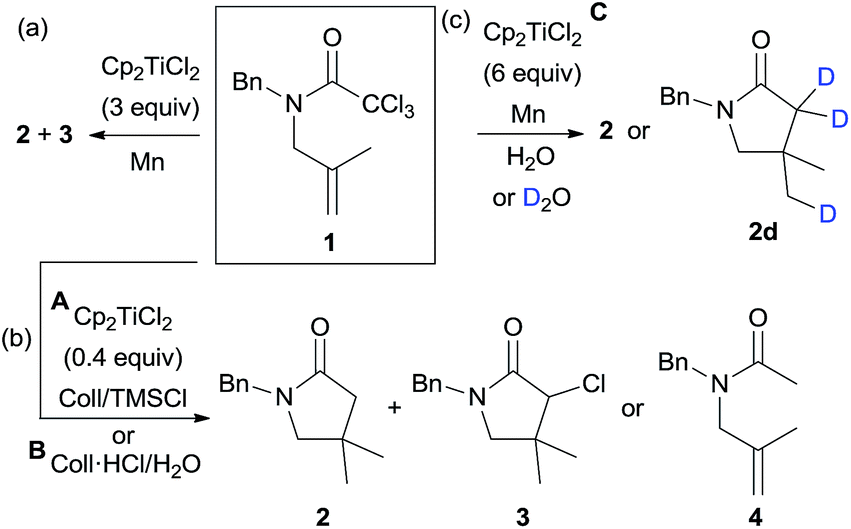 | ||
| Scheme 2 Cp2TiCl-mediated/catalysed cyclisations of trichloroacetamide 1. | ||
| Entry | Protocola | Eq. of Cp2TiCl2 | Yield (2 or 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio) :3 ratio) |
Yield (4) |
|---|---|---|---|---|
| a Protocol A: 8 eq. of Mn dust, 6 eq. of 2,4,6-collidine, and 4 eq. of TMSCl, 72 h. Protocol B: 8 eq. of Mn dust, 4 eq. of 2,4,6-collidine hydrochloride, and 10 eq. of H2O, 72 h. Protocol C: 12 eq. of Mn dust, and 10 eq. of H2O, 72 h. b 24 h of reaction. c 48 h of reaction. d 10 eq. of D2O were used. | ||||
| 1 | Ab | 0.2 | 58% (7:3) |
— |
| 2 | Ac | 0.2 | 62% (8:2) |
— |
| 3 | A | 0.2 | 90% (8:2) |
— |
| 4 | A | 0.4 | 91% (1:0) |
— |
| 5 | A | 0.6 | 63% (1:0) |
— |
| 6 | A | 0.8 | 51% (1:0) |
— |
| 7 | B | 0.4 | 0% | 79% |
| 8 | B | 0.6 | 0% | 81% |
| 9 | B | 0.8 | 0% | 88% |
| 10 | C | 6 | 36% (1:0) |
— |
| 11 | Cd | 6 | 30% (1:0) |
— |
Cp2TiCl-catalysed cyclisations of compound 1 yielded the corresponding γ-lactam 2 as the main product. In absence of water, the best result was obtained when 0.4 equiv. of Cp2TiCl was used after 72 h of reaction, which gave 2 in high yield (91%, see Table 1, entry 4). A mixture of 2 and 3 was obtained in a similar yield when less Cp2TiCl was employed (Table 1, entry 3). When the amount of catalyst was increased (see Table 1, entries 5 and 6), compound 2 was isolated in worse yields. This fact could be justified considering that with low amounts of catalyst (0.2 equiv., entries 1–3) the dehalogenation process is slow, resulting in a lower yield of 2 and substantial amounts of 3. Nevertheless, with more Cp2TiCl (0.6 or 0.8 equiv., entries 5–6), side reactions can take place. Additionally, the highly oxophilic Cp2TiCl complex could trap the dichloromethylcarbamoyl radical intermediate, yielding an inert enolate unable to continue the cyclisation reaction.7d
Taking into account that the use of TMSCl in these reaction conditions completely excludes the presence of adventitious water in the media, there must be an alternative source of hydrogen atoms. Newcomb24 and Cuerva23d have previously proposed that THF, when used as a solvent, is also able to act as a hydrogen-atom donor in Ti(III)-mediated processes.25 On the other hand, when the combination of Cp2TiCl and water was used as the hydrogen atom source, acyclic product 4 was detected (Scheme 2b, and Table 1, entries 7–9). This fact shows that under these reaction conditions, the reduction of the generated radicals is faster than the 5-exo-cyclisation. Increasing the amounts of Cp2TiCl to 6 equiv., we obtained product 2 (36%) (Table 1, entry 10). Under the same conditions, but using deuterium oxide instead of water, a trideuterated product 2d was obtained (Table 1, entry 11), with 46% deuterium incorporation,26 thus confirming that in this case the origin of the hydrogen atoms was via the titanocene(III) aqua-complex.23
Based on these results, the outcome of the reaction could be explained by the following mechanism (Scheme 3). The reaction begins with the dehalogenation between trichloroacetamide 1 and Cp2TiCl (generated from the reduction of Cp2TiCl2 with Mn dust) to yield radical I. Subsequently, this radical carries out a 5-exo-trig cyclisation, yielding cyclic intermediate II. The primary radical generated in this step is reduced by a hydrogen atom source, such as Ti(III)-aquacomplex or the solvent (THF), present in the reaction media, yielding lactam 5. Then, consecutive dehalogenation processes yield α-carbonyl radicals III and IV, which are also reduced, thus leading to the final lactam 2. The different titanocene(IV) species generated during the process are reintroduced in the catalytic cycle by the action of the regenerating agent and Mn dust, closing the catalytic cycle.
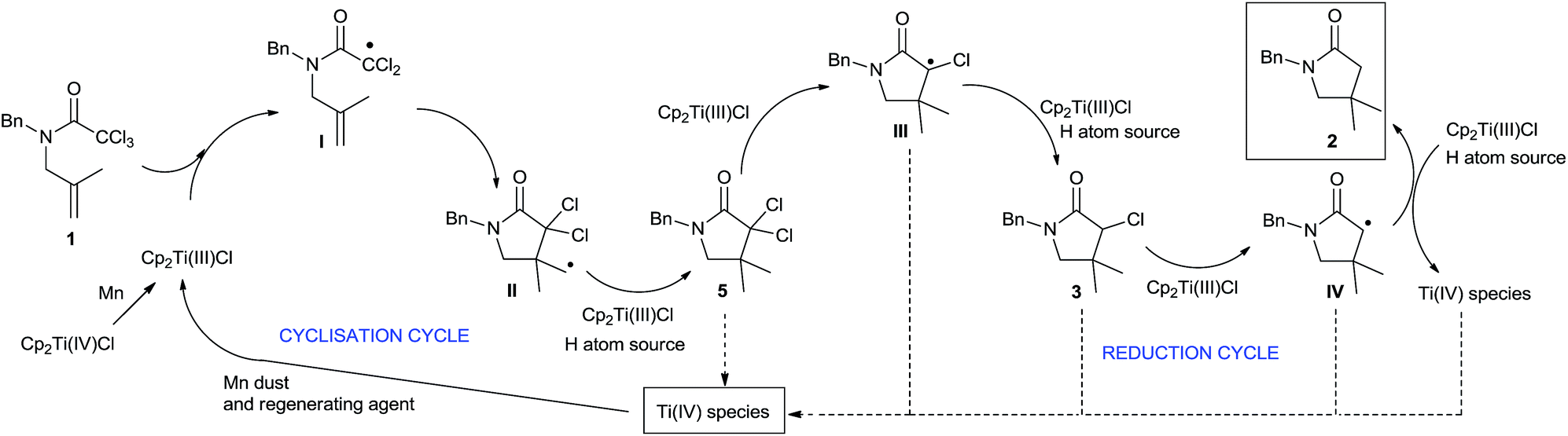 | ||
| Scheme 3 Proposed mechanism for titanocene(III)-catalysed radical cyclisation of trichloroacetamides. | ||
With these results in hand, we decided to extend the optimized reaction conditions to other substrates, including acyclic and cyclic compounds (Table 2).
| Entry | Acetamide | Product | Yield |
|---|---|---|---|
|
a 0.4 equiv. of Cp2TiCl, 8 equiv. of Mn dust, 6 equiv. of 2,4,6-collidine, and 4 equiv. of TMSCl, 72 h.
b 65:35 products ratio.
c 1:1 mixture of alkene and reduction products.
d 2 equiv. of Cp2TiCl, and 10 equiv. of H2O were used.
e 9:1 isomer ratio.
f 7:3 isomer ratio.
g 2:1 isomer ratio.
h 19% of monohalogenated derivative was also obtained.
i 1:1 isomer ratio.
|
|||
| 1 |
|
|
81%b |
| 2 |
|
|
75% |
| 3 |
|
|
70%c |
| 4 |
|
|
71%d |
| 5 |
|
|
44% |
| 6 |
|
|
58%e |
| 7 |
|
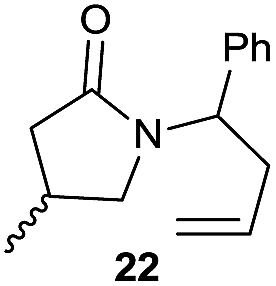
|
61%f |
| 8 |
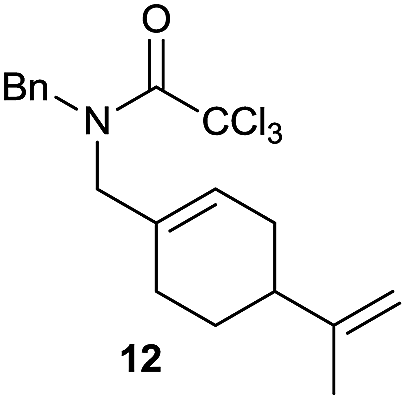
|
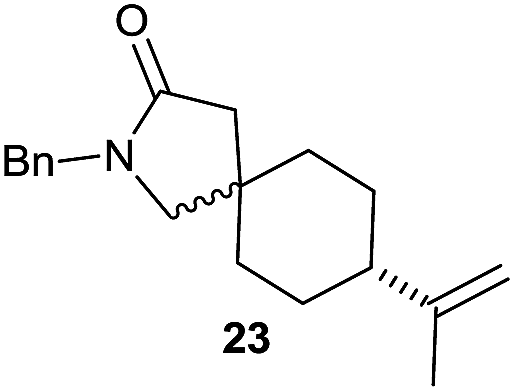
|
65%g |
| 9 |
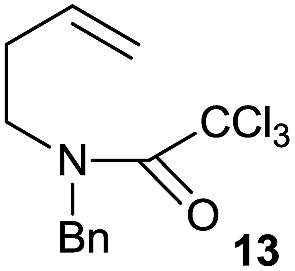
|
|
85% |
| 10 |
|
|
81%h |
| 11 |
|
|
48%i |
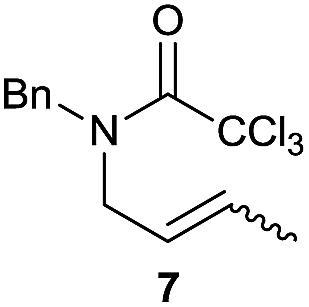
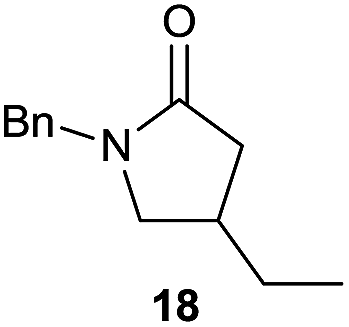
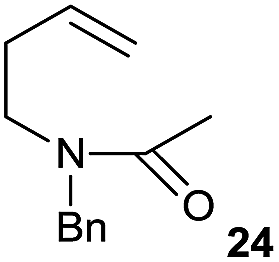
The cyclisation of trichloroacetamides 6–15 occurred efficiently,27 with moderate to high yields, providing straightforward access to a diversity of completely dehalogenated compounds. The cyclisation products mainly present a 5-membered ring (entries 1–8), although in some cases 6-membered rings were also obtained (entries 10–11). When both 5-exo-trig or 6-endo-trig cyclisations were possible in the same substrate (entry 7), we only obtained γ-lactam 22, derived from a 5-exo-trig process. This selectivity could be explained by the different rates of these cyclisations in radical processes.28 Moreover, the relative slowness of 6-endo-trig cyclisations compared with 5-exo-trig processes favours an early trapping of the intermediate radicals by titanocene species, avoiding and/or hindering the cyclisation processes. In fact, when trichloroacetamide 13 was submitted to our reaction conditions, we only recovered the corresponding dehalogenated acyclic compound 24 in good yield. On the other hand, these results also show that this reaction could be used as a mild and efficient procedure for complete dehalogenation of α-carbonyl polyhalogenated compounds.29
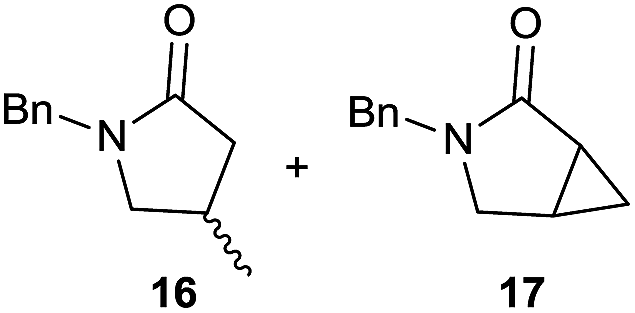
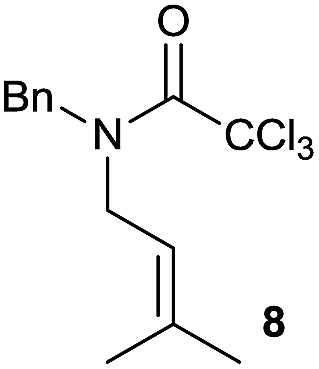
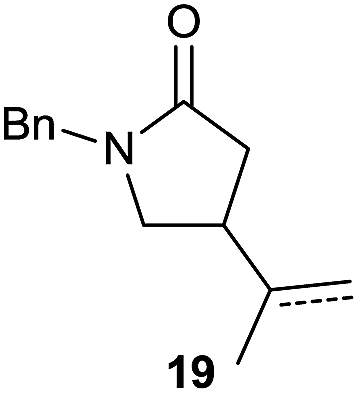
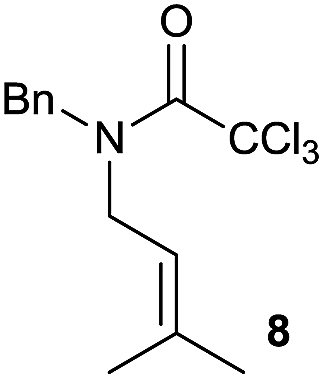
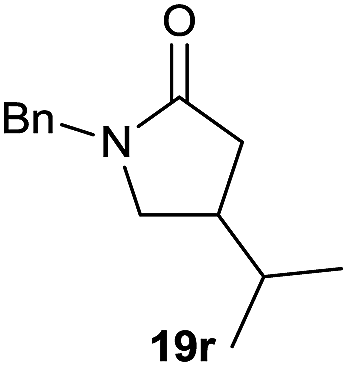
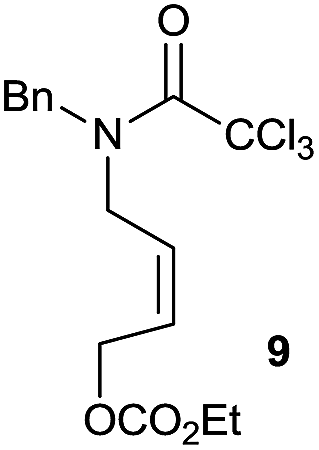
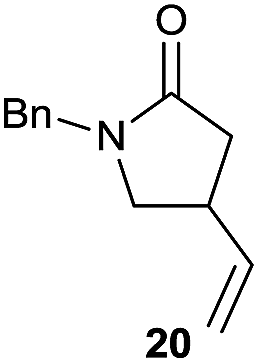
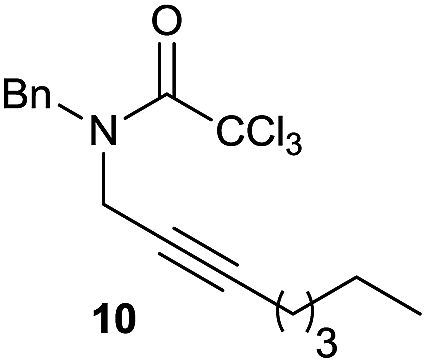
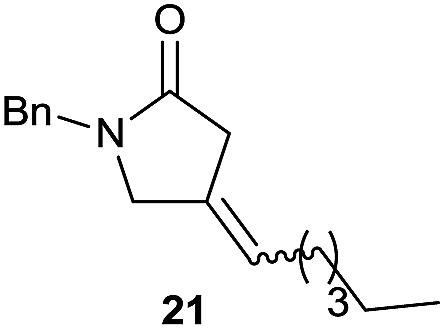
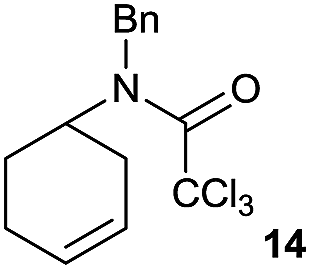
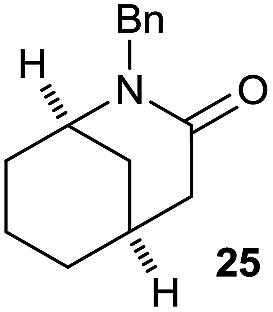
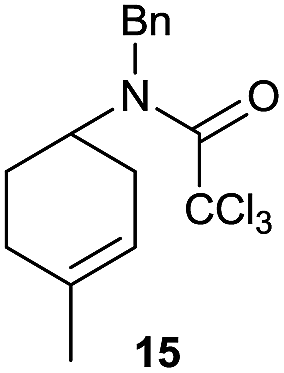
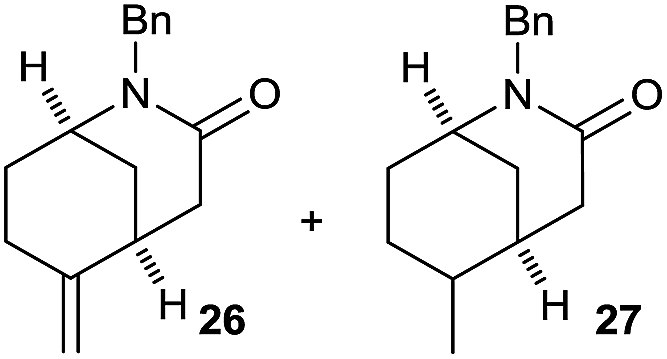
It is noteworthy that in the cyclisation of compound 6, 17 was also obtained (65:35 ratio with respect to 16). This bicyclic compound has been previously obtained as the main product in the Ru-catalysed radical cyclisation of a dichloro-derivative of 6.30 In our case, the formation of 17 implies the coexistence of two C-centred radicals, derived from dehalogenation and cyclisation steps. Cyclisation of 8 yielded a mixture of compounds 19 and 19r, which derive from two different oxidative and reductive ending processes, as we have previously described.23c The reduced 19r was prepared selectively, using a combination of 2 equiv. of Cp2TiCl and 10 equiv. of H2O. Cyclisation of compound 9 led to product 20 (entry 5), and subsequent removal of the carbonate group yielded an alkene. This termination step has been previously reported in several studies as being due to a Cp2TiCl-mediated radical fragmentation of β-carboxy radicals.6,7b,31 Our process also worked with alkynes as radical acceptors32 (entry 6), yielding the corresponding γ-lactam 21. The 6-exo-trig radical cyclisation of compounds 14–15 (entries 10–11) yielded morphans 25–27, thus constituting a new procedure to achieve this bridged azabicyclic scaffold33 through a titanocene(III)-based methodology.
To obtain deeper insight into the observed reactivity and selectivity of compounds 1 and 14, and the absence of cyclisation from compound 13, we performed DFT calculations on those structures,34 locating the transition states for all possible cyclisations and for the alternative hydrogen-atom transfer (HAT) process from THF (Scheme 4). The comparison of an intramolecular process (cyclisation) with an intermolecular one (HAT) is not straightforward, and should be done in terms of Gibbs free energy, but the large amount of THF available in the medium also allowed the utilization of enthalpies as representative values of the relative strength of the breaking and forming bonds. Our calculations confirm that γ-lactam derivatives are selectively formed through 5-exo processes, and also that in compounds containing one more carbon atom in the chain (13), the HAT process from THF becomes a competitive process. Initially, three transition structures were located from intermediate 1-rad, the radical species derived from compound 1. The structure lowest in energy corresponds to the 5-exo-trig cyclisation process (TS1, 3.4 kcal mol−1 at M06-2X level, Scheme 4a), favouring the formation of 5-membered rings, and the difference with the regioisomeric 6-endo-process (TS2, 8.9 kcal mol−1) is large enough to ensure the complete selectivity of the reaction. The HAT process between 1-rad and THF is also predicted to be higher in energy than TS1 (TS3, 7.5 kcal mol−1). The calculations at B3LYP level show higher absolute energy values, but similar trends. These results explain the favoured cyclisation and complete exo-selectivity shown in Table 2, entries 1–8.
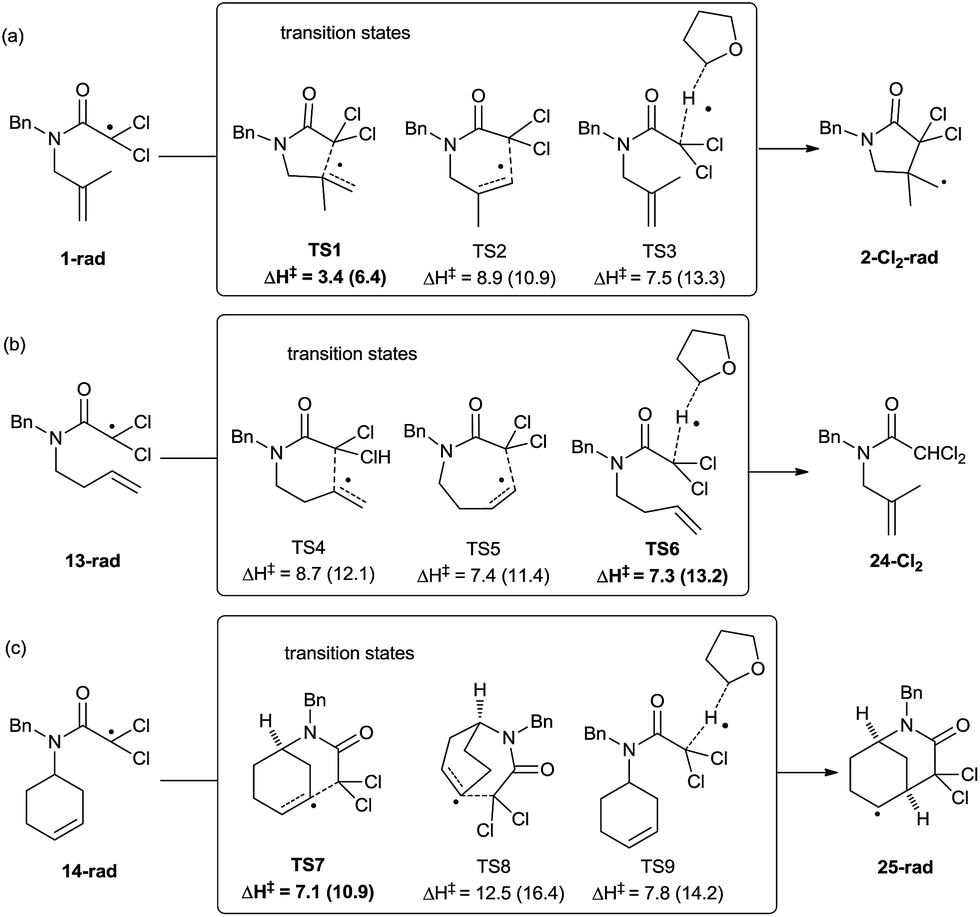 | ||
| Scheme 4 Enthalpy values computed for the transition states arising from 1-rad, 13-rad, and 14-rad at M06-2X/6-311+G(d,p) level of theory. Values in parenthesis correspond to B3LYP/6-311+G(d,p). | ||
When the homologous substrate 13-rad was computed, the energy values varied substantially from the previous case (Scheme 4b), and the endo approach (TS5, ΔH‡ = 7.4 kcal mol−1) became favoured over exo (TS4, ΔH‡ = 8.7 kcal mol−1) at both computational levels (M06-2X and B3LYP) by ca. 1 kcal mol−1. Even more interestingly, the hydrogen-atom abstraction from THF becomes competitive, presenting the lowest activation energy at M06-2X (TS6, 7.3 kcal mol−1). Obviously, the HAT process shows similar activation barriers for substrates 1 and 13 (compare the energies of TS3vs.TS6), but notably, 5-exo cyclisation in 1 would be much faster than the 6-exo process in 13. These values would explain the absence of cyclisation for compounds 13 and its conversion into 24 (Table 2, entry 9). The last two substrates in Table 2 (14 and 15) yield bicyclic adducts, through 6-exo cyclisations. In agreement with the experimental findings, TS7 presents the lowest activation values with the two functionals (ΔH‡ = 7.1–10.9 kcal mol−1, Scheme 4c), and its preference with respect to the HAT process increases slightly when compared with the previous substrate (13). Two opposite effects can explain the reactivity differences of Schemes 4b and c. Compound 14 is more prone to undergo cyclisation than 13 (TS7 is ca. 1.0–1.5 kcal mol−1 lower in energy than TS4) due to its lower flexibility and higher preorganization, and the HAT process from THF is ca. 1 kcal mol−1 less favoured (TS9vs.TS6) due to a higher steric hindrance in 14. Moreover, we also compared the transition state to form the bicyclo[3.2.2] compound (TS8), which is not energetically competitive (ΔH‡ = 12.5 kcal mol−1).
Conclusions
In summary, in this work we report trichloroacetamides as new suitable starting materials for titanocene(III)-catalysed reactions. We have applied this reactivity for the cyclisation of a variety of unsaturated trichloroacetamides to yield the corresponding γ-lactams, using simple and mild reaction conditions. This method also allowed the preparation of δ-lactams, but only when a favourable conformation was present in the starting materials. The procedure additionally allows the complete dehalogenation of trichloroacetamides, providing a mild and efficient alternative for the complete reduction of a variety of α-carbonyl polyhalogenated compounds.Acknowledgements
We thank the Regional Government of Andalucía (project P12-FQM-790), and MINECO (projects CTQ2014-53598, and CTQ2013-41338-P) for financial support.Notes and references
- M. L. H. Green and C. R. J. Lucas, J. Chem. Soc., Dalton Trans., 1972, 1000–1003 RSC.
- (a) W. A. Nugent and T. V. RajanBabu, J. Am. Chem. Soc., 1988, 110, 8561–8562 CrossRef CAS; (b) T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1989, 111, 4525–4527 CrossRef CAS; (c) T. V. RajanBabu, W. A. Nugent and M. S. Beattie, J. Am. Chem. Soc., 1990, 112, 6408–6409 CrossRef CAS; (d) T. V. Rajan-Babu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986–997 CrossRef CAS.
- (a) A. Gansäuer, H. Bluhm and M. Pierobon, J. Am. Chem. Soc., 1998, 120, 12849–12859 CrossRef; (b) A. Gansäuer, M. Pierobon and H. Bluhm, Angew. Chem., Int. Ed., 1998, 37, 101–103 CrossRef; (c) A. Gansäuer, T. Lauterbach, H. Bluhm and M. Noltenmeyer, Angew. Chem., Int. Ed., 1999, 38, 2909–2910 CrossRef; (d) A. Gansäuer, J. Chem. Soc., Chem. Commun., 1997, 457–458 RSC. For pinacol reactions, also see: (e) M. C. Bardena and J. Schwartz, J. Am. Chem. Soc., 1996, 118, 5484–5485 CrossRef.
- (a) J. Justicia, A. Rosales, E. Buñuel, J. L. Oller-López, M. Valdivia, A. Haidour, J. E. Oltra, A. F. Barrero, D. J. Cárdenas and J. M. Cuerva, Chem.–Eur. J., 2004, 10, 1778–1788 CrossRef CAS PubMed; (b) A. Rosales, J. L. Oller-López, J. Justicia, A. Gansäuer, J. E. Oltra and J. M. Cuerva, Chem. Commun., 2004, 2628–2629 RSC; (c) R. E. Estévez, M. Paradas, A. Millán, T. Jiménez, R. Robles, J. M. Cuerva and J. E. Oltra, J. Org. Chem., 2008, 73, 1616–1619 CrossRef PubMed.
- J. D. Parrish, D. R. Shelton and R. D. Little, Org. Lett., 2003, 5, 3615–3617 CrossRef CAS PubMed.
- S. P. Morcillo, D. Miguel, A. G. Campaña, L. Álvarez de Cienfuegos, J. Justicia and J. M. Cuerva, Org. Chem. Front., 2014, 1, 15–33 RSC.
- For a review, see: (a) J. Justicia, L. Álvarez de Cienfuegos, A. G. Campaña, D. Miguel, V. Jakoby, A. Gansäuer and J. M. Cuerva, Chem. Soc. Rev., 2011, 40, 3525 RSC. For some recent applications: (b) M. Yamaoka, A. Nakazaki and S. Kobayashi, Tetrahedron Lett., 2009, 50, 6764–6768 CrossRef CAS; (c) L. Shi, K. Meyer and M. F. Greaney, Angew. Chem., Int. Ed., 2010, 49, 9250–9253 CrossRef CAS PubMed; (d) T. Jiménez, S. P. Morcillo, A. Martín-Lasanta, D. Collado-Sanz, D. J. Cárdenas, A. Gansäuer, J. Justicia and J. M. Cuerva, Chem.–Eur. J., 2012, 18, 12825–12833 CrossRef PubMed; (e) J. Justicia, T. Jiménez, D. Miguel, R. Contreras-Montoya, R. Chahboun, E. Álvarez-Manzaneda, D. Collado-Sanz, D. J. Cárdenas and J. M. Cuerva, Chem.–Eur. J., 2013, 19, 14484–14495 CrossRef CAS PubMed; (f) S. P. Morcillo, D. Miguel, S. Resa, A. Martín-Lasanta, A. Millán, D. Choquesillo-Lazarte, J. M. García-Ruiz, A. J. Mota, J. Justicia and J. M. Cuerva, J. Am. Chem. Soc., 2014, 136, 6943–6951 CrossRef CAS PubMed.
- (a) A. G. Campaña, B. Bazdi, N. Fuentes, R. Robles, J. M. Cuerva, J. E. Oltra, S. Porcel and A. M. Echavarren, Angew. Chem., Int. Ed., 2008, 47, 7515–7519 CrossRef PubMed; (b) A. Millán, A. G. Campaña, B. Bazdi, D. Miguel, L. Álvarez de Cienfuegos, A. M. Echavarren and J. M. Cuerva, Chem.–Eur. J., 2011, 17, 3985–3994 CrossRef PubMed; (c) A. Millán, L. Álvarez de Cienfuegos, A. Martín-Lasanta, A. G. Campaña and J. M. Cuerva, Adv. Synth. Catal., 2011, 353, 73–78 CrossRef; (d) A. Millán, A. Martín-Lasanta, D. Miguel, L. Álvarez de Cienfuegos and J. M. Cuerva, Chem. Commun., 2011, 47, 10470–10472 RSC; (e) A. Millán, L. Álvarez de Cienfuegos, D. Miguel, A. G. Campaña and J. M. Cuerva, Org. Lett., 2012, 14, 5984–5987 CrossRef PubMed.
- A. Rosales, J. Muñoz-Bascón, C. López-Sánchez, M. Álvarez-Corral, M. Muñoz-Dorado, I. Rodríguez-García and J. E. Oltra, J. Org. Chem., 2012, 77, 4171–4176 CrossRef CAS PubMed.
- J. Streuff, M. Feurer, P. Bichovski, G. Frey and U. Gellrich, Angew. Chem., Int. Ed., 2012, 51, 8661–8664 CrossRef CAS PubMed.
- G. Frey, H.-T. Luu, P. Bichovski, M. Feurer and J. Streuff, Angew. Chem., Int. Ed., 2013, 52, 7131–7134 CrossRef CAS PubMed.
- X. Zheng, J. He, H.-H. Li, A. Wang, X.-J. Dai, A.-E. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2015, 54, 13739–13742 CrossRef CAS PubMed.
- X. Zheng, X.-J. Dai, H.-Q. Yuan, C.-X. Ye, J. Ma and P.-Q. Huang, Angew. Chem., Int. Ed., 2013, 52, 3494–3498 CrossRef CAS PubMed.
- For pioneering works, see: (a) H. Nagashima, H. Wakamatsu and K. Itoh, J. Chem. Soc., Chem. Commun., 1984, 652–653 RSC; (b) Y. Hirai, A. Hagiwara, T. Terada and T. Yamazaki, Chem. Lett., 1987, 2417–2418 CrossRef CAS; (c) H. Nagashima, N. Ozaki, M. Ishii, K. Seki, M. Washiyama and K. Itoh, J. Org. Chem., 1993, 58, 464–670 CrossRef CAS.
- (a) J. Quirante, C. Escolano, A. Merino and J. Bonjoch, J. Org. Chem., 1998, 63, 968–976 CrossRef CAS; (b) Q. Li, G. Li, S. Ma, P. Feng and Y. Shi, Org. Lett., 2013, 15, 2601–2603 CrossRef CAS PubMed; (c) F. Diaba, A. Martínez-Laporta, G. Coussanes, I. Fernández and J. Bonjoch, Tetrahedron, 2015, 71, 3642–3651 CrossRef CAS.
- For the use of radical processes in the total synthesis of natural products, see: J. Bonjoch, B. Bradshaw and F. Diaba, in Stereoselective Synthesis of Drugs and Natural Products, ed. V. Andrushko and N. Andrushko, Wiley-VCH, New York, 2013, pp. 733–768 Search PubMed.
- (a) S. Iwamatsu, H. Kondo, K. Matsubara and H. Nagashima, Tetrahedron, 1999, 55, 1687–1706 CrossRef CAS; (b) A. J. Clark, Chem. Soc. Rev., 2002, 31, 1–11 RSC; (c) M. Pattarozzi, F. Roncaglia, V. Giangiordano, P. Davoli, F. Prati and F. Ghelfi, Synthesis, 2010, 694–700 CrossRef CAS; (d) Y. Motoyama, K. Kamo, A. Yuasa and H. Nagashima, Chem. Commun., 2010, 46, 2256–2258 RSC; (e) W. T. Eckenhoff and T. Pintauer, Catal. Rev.: Sci. Eng., 2010, 52, 1–59 CrossRef CAS; (f) F. Diaba, A. Martínez-Laporta, J. Bonjoch, A. Pereira, J. M. Muñoz-Molina, T. R. Belderraín and P. J. Pérez, Chem. Commun., 2012, 50, 8799–8801 RSC.
- (a) B. A. Seigal, C. Fajardo and M. L. Snapper, J. Am. Chem. Soc., 2005, 127, 16329–16332 CrossRef CAS PubMed; (b) C. D. Edlin, J. Faulkner and P. Quayle, Tetrahedron Lett., 2006, 47, 1145–1151 CrossRef CAS; (c) F. I. McGonagle, L. Brown, A. Cooke and A. Sutherland, Org. Biomol. Chem., 2010, 8, 3418–3425 RSC; (d) F. Diaba, A. Martínez-Laporta and J. Bonjoch, J. Org. Chem., 2014, 79, 9365–9372 CrossRef CAS PubMed.
- J. Cassayre, B. Quiclet-sire, J.-B. Saunier and S. Z. Zard, Tetrahedron, 1998, 54, 1029–1040 CrossRef CAS.
- M. Benedetti, L. Forti, F. Ghelfi, U. M. Pagnoni and R. Ronzoni, Tetrahedron, 1997, 53, 14031–14042 CrossRef CAS.
- J. Quirante, C. Escolano, M. Massot and J. Bonjoch, Tetrahedron, 1997, 53, 1391–1402 CrossRef CAS.
- J. Quirante, C. Escolano, F. Diaba and J. Bonjoch, J. Chem. Soc., Perkin Trans. 1, 1999, 1157–1162 RSC.
- (a) J. M. Cuerva, A. G. Campaña, J. Justicia, A. Rosales, J. L. Oller–López, R. Robles, D. J. Cárdenas, E. Buñuel and J. E. Oltra, Angew. Chem., Int. Ed., 2006, 45, 5522–5526 CrossRef CAS PubMed; (b) M. Paradas, A. G. Campaña, M. L. Marcos, J. Justicia, A. Haidour, R. Robles, D. J. Cárdenas, J. E. Oltra and J. M. Cuerva, Dalton Trans., 2010, 39, 8796–8800 RSC; (c) T. Jiménez, A. G. Campaña, B. Bazdi, M. Paradas, D. Arráez-Román, A. Segura-Carretero, A. Fernández-Gutiérrez, J. E. Oltra, R. Robles, J. Justicia and J. M. Cuerva, Eur. J. Org. Chem., 2010, 4288–4295 CrossRef; (d) M. Paradas, A. G. Campaña, T. Jiménez, R. Robles, J. E. Oltra, E. Buñuel, J. Justicia, D. J. Cárdenas and J. M. Cuerva, J. Am. Chem. Soc., 2010, 132, 12748–12756 CrossRef CAS PubMed; (e) A. Gansäuer, M. Behlendorf, A. Cangoenuel, C. Kube, J. M. Cuerva, J. Friedrich and M. van Gastel, Angew. Chem., Int. Ed., 2012, 51, 3266–3270 CrossRef PubMed; (f) Y.-Q. Zhang, V. Jakoby, K. Stainer, A. Schmer, S. Klare, M. Bauer, S. Grimme, J. M. Cuerva and A. Gansäuer, Angew. Chem., Int. Ed., 2016, 55, 1523–1526 CrossRef CAS PubMed. For other precedent of water acting as HAT reagent: (g) D. A. Spiegel, K. B. Wiberg, L. N. Schacherer, M. R. Medeiros and J. L. Wood, J. Am. Chem. Soc., 2005, 127, 12513–12515 CrossRef CAS PubMed.
- J. Jin and M. Newcomb, J. Org. Chem., 2008, 73, 7901–7905 CrossRef CAS PubMed.
- To check this fact, we performed cyclisation of 1 in the presence of THF-d8 as the solvent, which yielded a complex mixture of products. The high isotopic effect of THF-d8 avoids the deuterium atom transfer to the radical, driving the reaction towards non-deuterated compounds.
- 37% of dideuterated compound was also observed in the mass spectra.
- The reaction using monohaloacetamide (Cl or Br) analogs to 6 only gave the uncyclised reduced acetamide.
- A. L. J. Beckwith, C. J. Easton and A. K. Serelis, J. Chem. Soc., Chem. Commun., 1980, 482–483 RSC.
- For other methodologies, see: A. J. Fry, Reduction of α-substituted carbonyl compounds –CX–CO– to carbonyl compounds –CH–CO–, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Elsevier, Oxford, 1991, vol. 8, pp. 983–995 Search PubMed.
- For formation of 17 and related compounds in Ru-catalysed radical cyclisations from haloacetamide analogs of 6, see: K. Thomes, G. Kiefer, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2009, 48, 8115–8119 CrossRef PubMed.
- For other recent example, see: I. R. Márquez, A. Millán, A. G. Campaña and J. M. Cuerva, Org. Chem. Front., 2014, 1, 373–381 RSC.
- A. Gansäuer, M. Otte and L. Shi, J. Am. Chem. Soc., 2011, 133, 416–417 CrossRef PubMed.
- J. Bonjoch, F. Diaba and B. Bradshaw, Synthesis, 2011, 993–1018 CrossRef CAS.
- See ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data. Computational details. Copies of 1H NMR and 13C NMR spectra of new compounds. See DOI: 10.1039/c6ra12180b |
| This journal is © The Royal Society of Chemistry 2016 |