DOI:
10.1039/C6RA11937A
(Paper)
RSC Adv., 2016,
6, 75440-75448
Novel environmentally friendly waterborne polyurethane/hollow Ni0.3Zn0.5Fe2O4 nanocomposite films: superior magnetic and mechanical properties
Received
8th May 2016
, Accepted 1st August 2016
First published on 1st August 2016
Abstract
Superparamagnetic nanocomposite films based on waterborne polyurethane and hollow Ni0.3Zn0.5Fe2O4 nanospheres (WPU/h-NZFO) were synthesized via an in situ polymerization technique. The h-NZFO nanoparticles were functionalized by vinyltriethoxysilane (VTES) to facilitate the dispersion and compatibility with the WPU matrix. The modified h-NZFO (h-NZFO-VTES), combined with the polymers through covalent bonds during the UV-curing process, acted as an effective cross-linker. The morphology, microstructure, thermal behavior, solvent resistance, mechanical and magnetic properties of the WPU/h-NZFO nanocomposites were investigated. It was found that functionalized h-NZFO nanoparticles could largely promote the emulsion stability, thermal stability, solvent resistance, mechanical and magnetic properties of WPU polymers. Importantly, the resulting WPU/h-NZFO films possess superior magnetic (24.2 emu cm−3) and mechanical properties, providing a promising route to prepare environmentally friendly WPU-based nanocomposites with high performance for practical applications, especially in microwave absorption fields.
1. Introduction
In recent decades, the structures and performances of film composites consisting of inorganic particles dispersed in different polymer matrices have been widely investigated both for fundamental reasons and practical applications.1–4 In this context, various inorganic particles have been used as fillers in different polymer matrices to form nanocomposites, such as SiO2, Fe3O4, TiO2, Au, MWCNTs, and attapulgite.5–10 The results showed that the addition of such inorganic fillers not only improved their physical and chemical properties, such as flame-retarding property, solvent resistance, mechanical properties etc.,11–13 but also endowed polymers with other special functionality, including electricity, optical properties, conductivity and bio-catalysis.14–17 WPU is a newly-developed branch of polyurethane systems (PUs) with a range of new physical and chemical properties. Water was used as solvent instead of traditional organic solvent, namely the polyurethane chains are dispersed in water, which reduces the release of volatile organic compounds (VOC). Therefore, WPU makes a great contribution to environmental protection.18 Meanwhile, compared other polymer materials, waterborne polyurethane possess many excellent advantages, such as abrasion resistance, flexibility, high hardness, superior process ability, non-toxicity etc.19,20 Therefore, they have been widely used in adhesives and coating for leather, textile, glass, and fiber paper.21–24 However, the inferior properties of WPU, including weak mechanical strength, poor solvent resistance and low thermal stability etc., hindered its wide application. Some measures have been taken to address this issue, for example, improved cross-linking density, added kinds of inorganic nanoparticles to WPU matrix.25,26 Nevertheless, in most of previous works, the WPU nanocomposites with the addition of inorganic particles were prepared through simple mechanical mixing, and the inorganic couldn't homogeneously dispersed in the organic phase matrix for the agglomeration of inorganic nanoparticles, this had a great negative effect on performances of final composite materials. Moreover, inorganic nanometer particles have been widely introduced into polymers only to improve the physical and mechanical properties of polymer materials without other special functionality.27,28
Ni–Zn ferrite is a kind of spinel ferrite and has been intensively investigated due to its high Curie temperature, high resistivity, low dielectric losses, and excellent microwave-absorbing properties.29 Based on above advantages, Ni–Zn ferrite has been used as filler, especially as functional particle adding in various polymer matrices to form nanocomposites, and have a wide application in microwave absorption materials, biocatalysis, high-density information storage fields.30,31 However, as far as we know, there are only a fewer papers reported Ni–Zn ferrite used as filler in WPU matrix to investigate its magnetic properties for its poor storage stability of WPU/Ni–Zn ferrite composite emulsion. Ni–Zn ferrites show higher density than common inorganic nanoparticles, they easily settle down in the WPU emulsion and separate from the organic phase, leading to phase separation, which made it difficult to prepare final WPU/Ni–Zn ferrite nanocomposites. The nanocomposites based on WPU and Ni–Zn ferrite not only present good physical properties, but also possess excellent magnetic properties, and they would hopefully have a wide application in microwave absorption. For example, the nanocomposites can be coated on aircrafts and ships surface to reduce the sensitivity of the radar, and also widely used in anechoic chambers etc.32
In the present work, a series of WPU/h-NZFO nanocomposites with excellent magnetic properties have been successfully prepared by in situ polymerization method. The hollow Ni0.3Zn0.5Fe2O4 nanoparticles modified with VTES were combined with the WPU matrix to improve properties of polymer matrix. The h-NZFO-VTES contained plenty of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds which would participate in radical polymerization reaction in UV-curing process. In this way, the h-NZFO-VTES would link with WPU matrix through covalent bonds instead of conventional physical mixing. Moreover, h-NZFO-VTES acted as effective cross-linker in the reaction system, which would contribute to better physical properties of nanocomposites.
C bonds which would participate in radical polymerization reaction in UV-curing process. In this way, the h-NZFO-VTES would link with WPU matrix through covalent bonds instead of conventional physical mixing. Moreover, h-NZFO-VTES acted as effective cross-linker in the reaction system, which would contribute to better physical properties of nanocomposites.
2. Experimental section
2.1 Materials
Triethylamine (TEA), zinc chloride (ZnCl2), and ammonium acetate (NH4Ac) were purchased from Tianjin Guangfu Technology Development Co. LTD (China); poly(propylene glycol) (PPG-2000) and ferric chloride hexahydrate (FeCl3·6H2O) were obtained from Sinopharm Chemical Reagent Co., Ltd (China). Nickel chloride hexahydrate (NiCl2·6H2O) was purchased from Tianjin Kermel Chemical Reagent Co., Ltd (China). Vinyltriethoxysilane (VTES) was obtained from Cheng Du Ke Long Chemical Co, Ltd (China). Isophorone diisocyanate and ethylene glycol (IPDI) were supplied by Shanghai Chemical Reagents Corp. (China); dimethylol propionic acid (DMPA) was purchased from Aldrich Chemical (China); hydroxyethyl methacrylate (HEMA) was obtained from Tianjin Institute of chemical reagents (China). Dibutyltin dilaurate (DBTDL) was purchased from Tianjin Kaixin Chemical Co. Ltd (China); Irgacure 2959 was obtained from Sigma-Aldrich Co. Ltd (China). DMPA was dried at 40 °C for 24 h before use. Acetone and toluene were dehydrated via 4A MS before use. All reagents were of analytical purity.
2.2 Preparation of h-NZFO-VTES nanoparticles
The Ni0.3Zn0.5Fe2O4 nanoparticle was prepared according to our previous work with little modification.33 FeCl3·6H2O (1.083 g), ZnCl2 (0.191 g), NiCl2·6H2O (0.143 g) and PEG-6000 (6.30 g) were dissolved in 70 mL of ethylene glycol under mechanical stirring to form a homogeneous solution. Then, 7.71 g NH4Ac was added to above system with constant stirring for 40 min. The homogeneous solution was transferred into a 100 mL teflon-lined stainless steel autoclave and was calcined at 160 °C for 10 h. After cooled down to room temperature naturally, the mixture was washed with water and ethanol several times, respectively. After that, the mixture was dried under vacuum at 40 °C for 24 h. To functionalized h-NZFO nanoparticle with double bond, the h-NZFO-VTES was fabricated as follows: 1 g h-NZFO powder was dissolved in 200 mL solvent (vethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :vH2O = 4:1) under mechanical stirring and maintained ultrasonic for 30 min. Then, 5 mL of ammonia and 8 mL of vinyltriethoxysilane were added to the solution. The reaction was carried out at 60 °C for 12 h under N2 protection. Finally, the product was washed with water and ethanol in sequence and dried under vacuum at 40 °C for 24 h.
:vH2O = 4:1) under mechanical stirring and maintained ultrasonic for 30 min. Then, 5 mL of ammonia and 8 mL of vinyltriethoxysilane were added to the solution. The reaction was carried out at 60 °C for 12 h under N2 protection. Finally, the product was washed with water and ethanol in sequence and dried under vacuum at 40 °C for 24 h.
2.3 Synthesis of WPU/h-NZFO nanocomposites
The WPU/h-NZFO-VTES was prepared according to following procedure. Firstly, IPDI (4.45 g), PPG-2000 (10.00 g) and DMPA (0.67 g) were added in a 100 mL dry flask equipped with a condenser, N2 inlet and mechanical stirring. Then, 5–6 drops of DBTDL were dropped into and the mixture was reacted at 80 °C for 3 h. Subsequently, the system was cooled to 60 °C, followed by the addition of HEMA (2.60 g) and reacted for 2 h under this condition. A stoichiometric ratio of h-NZFO-VTES was introduced to reaction and kept ultrasonic for 1 h. Later, the mixture was reacted at 60 °C for 2 h. TEA was added to emulsify the dispersions and stirred for 30 min. At last, a certain amount of water was added to emulsify above neutralized dispersion and stirred for 40 min at 40 °C. The solid content of resulting dispersion was 30%. The WPU/h-NZFO nanocomposite film was prepared by mixing preweighted above emulsion with Irgacure 2959 (3 wt%) under mechanical stirring. Subsequently, the composite latex was spreaded on a cleaned Teflon disk and placed under a UV light (wavelength of 295 nm, power of 1000 W) to cure for 4 min. The procedure is shown in Scheme 1. The nanocomposite films with h-NZFO-VTES mass fraction of 0, 0.5, 1.0, 1.5 and 2.0 were marked as WPU, WPU/h-NZFO-0.5, WPU/h-NZFO-1.0, WPU/h-NZFO-1.5 and WPU/h-NZFO-2.0, respectively.
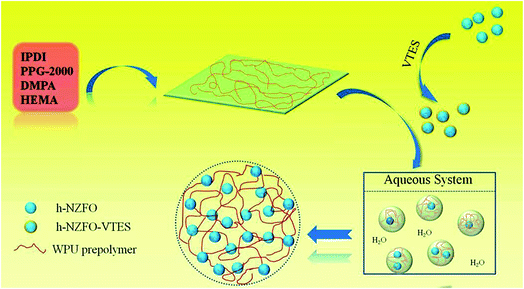 |
| | Scheme 1 Schematic illustration of the synthesis process of WPU/h-NZFO nanocomposites. | |
2.4 Characterization
The morphology of product was investigated using FEI Tecnai G20 transmission electron microscopy. The scanning electron microscopy (SEM, JSM-6380Lv, JEOL, Japan; JSM-6701F, JEOL, Japan) was used to evaluate the morphology and dispersion of h-NZFO-VTES nanoparticles in the matrix. The crystal identification of fine powders and films were characterized by X-ray powder diffraction (XRD, Rigaku D/MAX-2400 X-ray diffractometer with Ni-filtered Cu Kα radiation). Fourier transform infrared (FTIR) spectra (American Nicolet Corp. Model 170-SX) were conducted to investigate the microstructure of products by KBr pelleting technique. The particle size and distribution of films were identified using dynamic light scattering (DLS) (Zeta-plus, Brookehaven Instruments). TGA curves were obtained by using a thermogravimetric analyzer from TA Instrument (NETZSCH STA 449C) under a stable N2 flow. DMA was performed with the Rheometric Q800 DMA (Mettler-Toledo) from −50 to 200 °C at a heating rate of 3 °C min−1, and the specimens used for the test were of dimensions 9 mm × 4 mm × 0.4 mm. The magnetic properties of samples were conducted on vibrating sample magnetism (VSM, LAKESHORE-7304, USA). The mechanical properties were measured on an Instron 1122 tensile apparatus according to GB1040-79 at a drawing rate of 30 mm min−1 at 20 °C.
The WPU/h-NZFO films were cut into the size of 2 cm × 2 cm, then was extracted with acetone after drying the sample for 48 h at room temperature. After extraction with acetone for 24 h, the film was dried under vacuum for 24 h and the gel content was calculated as following formula:
where “
M” is the initial mass of dry film, and “
m” is the mass of film after drying under vacuum.
The water (toluene) absorption of film sample was carried out by immersing film in water (toluene) for a certain period of time. The water (toluene) absorption was calculated using following formula:
where “
ma” is the initial mass of film, and “
mb” is the mass of film after immersing in water (toluene) for a specified time interval.
The storage stability of the polymer emulsion was determined according to stable time (time before emulsion stratification) and was performed as follows: standing the WPU/h-NZFO emulsions under natural condition without a slight movement and observing the appearance of emulsions.
3. Results and discussion
3.1 Elemental analysis
Table 1 shows the data for elemental analysis of h-NZFO and h-NZFO-VTES particles. The N element content of both h-NZFO and h-NZFO-VTES particles were nearly 0.14%, according with the sample element composition. The C and H mass fraction of pristine h-NZFO are 1.92% and 0.27%, respectively, and increased to 2.54% and 0.33% for h-NZFO-VTES, indicating that h-NZFO was successfully modified with VTES.
Table 1 Elemental analysis of h-NZFO and h-NZFO-VTES
| Designation |
N (%) |
C (%) |
H (%) |
| h-NZFO |
0.14 |
1.92 |
0.27 |
| h-NZFO-VTES |
0.13 |
2.54 |
0.33 |
3.2 Morphology and microstructure of h-NZFO and WPU/h-NZFO nanocomposite films
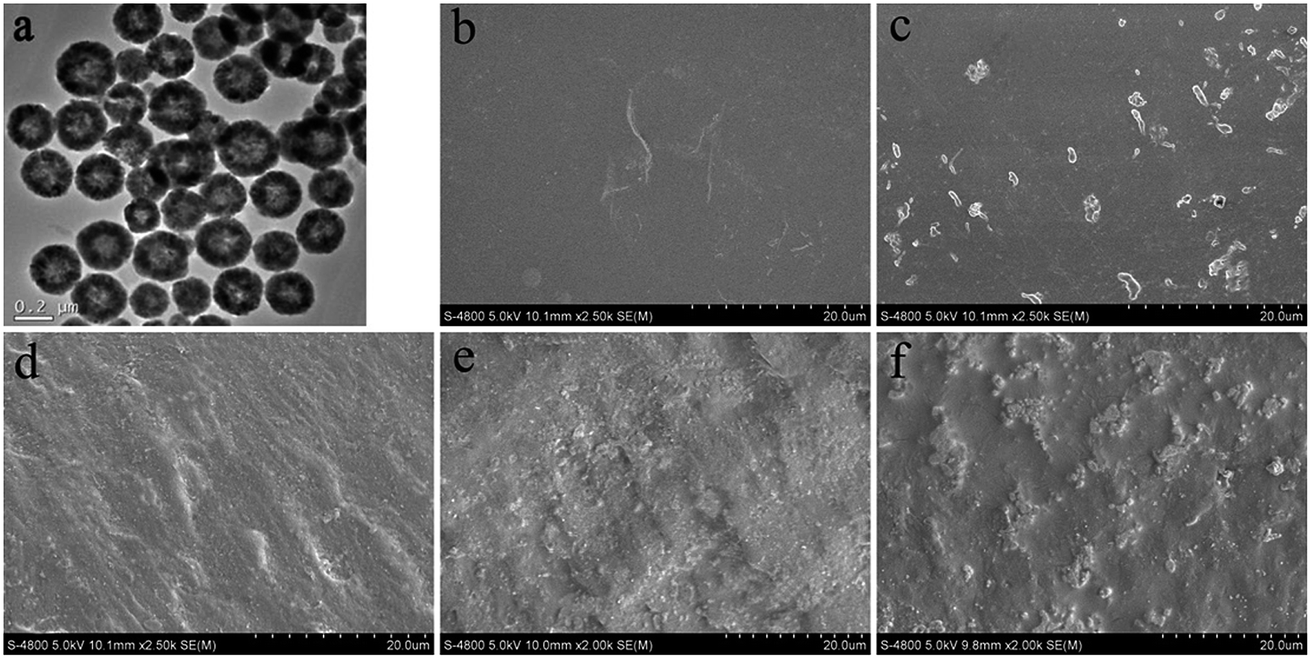
TEM and SEM tests were carried out to investigate the morphologies and distribution of products and were shown in Fig. 1. The h-NZFO particles (Fig. 1a) are spherical with diameter of 195 nm and all particles display hollow structure. Fig. 1b–f are SEM photographs of fractured surface of pure WPU and WPU/h-NZFO nanocomposite films. The fractured section of pure WPU shows a glassy and smooth surface (Fig. 1b). Compared with pristine WPU, the films with h-NZFO-VTES nanoparticles presented much rougher surface. Fig. 1c and d were nanocomposite films with pristine h-NZFO and h-NZFO-VTES content of 1.0%, respectively. It was observed that h-NZFO-VTES nanoparticles are homogeneously dispersed in the WPU matrix, while pristine h-NZFO nanoparticles almost aggregate together and are exposed on the polymer matrix surface. Fig. 1e and f are SEM images of WPU/h-NZFO-1.5 and WPU/h-NZFO-2.0, respectively. It was observed that the h-NZFO-VTES nanoparticles were well adhered with WPU matrix when the h-NZFO-VTES content is 1.5%. This indicated that the functionalized h-NZFO nanoparticles improved dispersion of the nanocomposites. However, obvious aggregation occurred in some area of WPU/h-NZFO-2.0 sample, which indicates that the uniform distribution of inorganic particles in matrix had a great effect on performances of final films.34
 |
| | Fig. 1 The TEM image of h-NZFO nanoparticles (a), the SEM photographs of fractured section of pure WPU (b), WPU/pristine h-NZFO-1.0 (c), WPU/h-NZFO nanocomposites with h-NZFO-VTES content is 1.0% (d) 1.5% (e) and 2.0% (f). | |
As shown in Fig. 2, the crystal structure of samples was examined by X-ray diffraction. From h-NZFO, several characteristic peaks of Ni–Zn ferrite occurred at 2θ = 18.1°, 30.3°, 35.2°, 43.1°, 53.6°, 56.7°, 62.6°, which match well with the single crystalline nickel zinc ferrite (JCPDS card no. 019-0629). The same diffraction peaks were observed in h-NZFO-VTES and WPU/h-NZFO-1.0. However, the peaks' intensity of h-NZFO-VTES became weaker compared with pristine h-NZFO particles due to interaction formed between h-NZFO and VTES. Moreover, the peak occurred between 15° and 25° in WPU/h-NZFO-1.0 should be attributed to the orderly arrangement of hard segment of WPU polymers.35
 |
| | Fig. 2 XRD patterns of h-NZFO (a), h-NZFO-VTES, (b) and WPU/h-NZFO-1.0 nanocomposite (c). | |
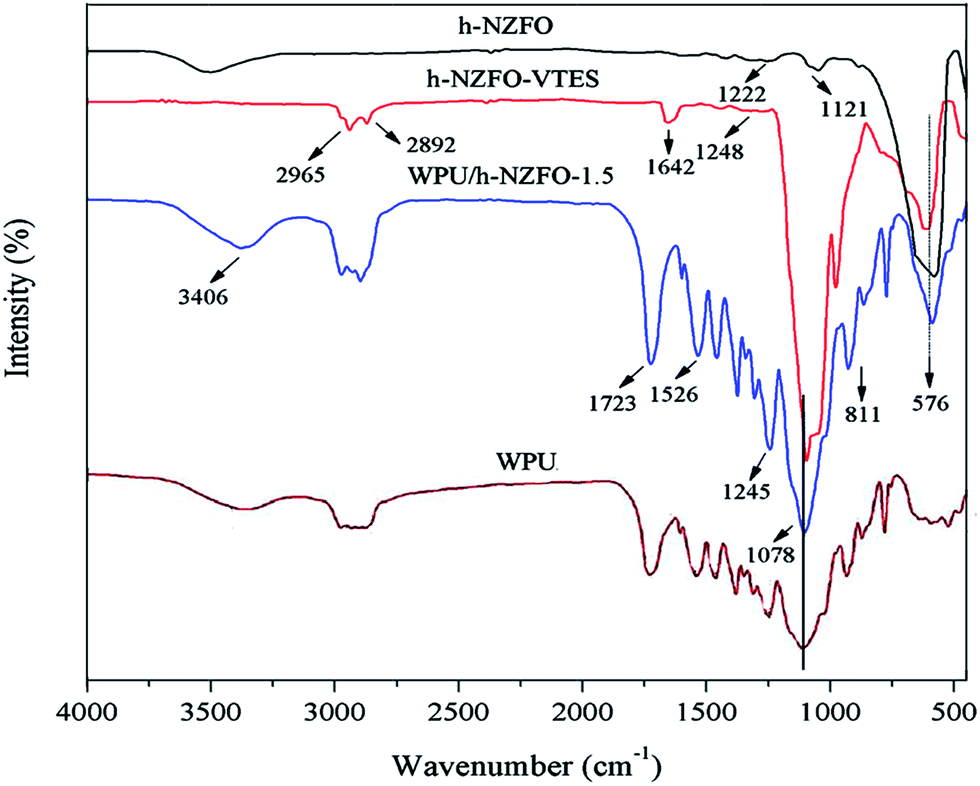
The chemical structures of samples were characterized by Fourier transform infrared (FTIR) spectra. Fig. 3 shows the FT-IR spectra of h-NZFO, h-NZFO-VTES, WPU, and WPU/h-NZFO-1.5. Form the spectrum of h-NZFO, a broad peak at 3440 cm−1 is assigned to –OH stretching. The band at around 576 cm−1 is attributed to the Fe–O bond stretching. In addition, the peaks in h-NZFO at 1222 and 1121 cm−1 are owing to the vibration of the C–O and C–H groups of the surfactant.36 Compared to h-NZFO, several new peaks were detected in h-NZFO-VTES. The peaks at 2965 and 2892 cm−1 are assigned to asymmetrical and symmetrical stretching peaks of –CH3 and –CH2 groups of VTES grafted to the surface of h-NZFO. The newly peaks occurring at 1078 and 811 cm−1 corresponded to the asymmetrical and symmetrical stretching vibration of Si–O bonds.5 The weak bond at 1248 cm−1 could be attributed to C–O bond. Meanwhile, the characteristic stretching vibration peak of CC bond is clearly observed at 1642 cm−1. Above analysis indicated that h-NZFO particle was successfully modified by VTES.
 |
| | Fig. 3 FT-IR spectra of h-NZFO, h-NZFO-VTES, WPU, and WPU/h-NZFO-1.5 nanocomposite. | |
In WPU and WPU/h-NZFO-1.5 spectra, a broad band at around 3406 cm−1 is the stretching peak of N–H bonds. The peaks at 1723 and 1528 cm−1 are attributed to the stretching vibration of CO and the bending vibration of N–H bond of –NH–CO–O–.10 The stretching peak of C–O bond is also detected at 1245 cm−1. Moreover, the peaks of –CH3 and –CH2 were strengthened. Additionally, compared to h-NZFO-VTES, the characteristic peak of CC bond wasn't observed in WPU/h-NZFO-1.5 nanocomposite, which implied that CC bonds of h-NZFO-VTES have participated in polymerization reaction during UV-curing process. All results indicated that the WPU/h-NZFO nanocomposite has been successfully obtained.
3.3 Size distribution and morphology of WPU and WPU/h-NZFO emulsions
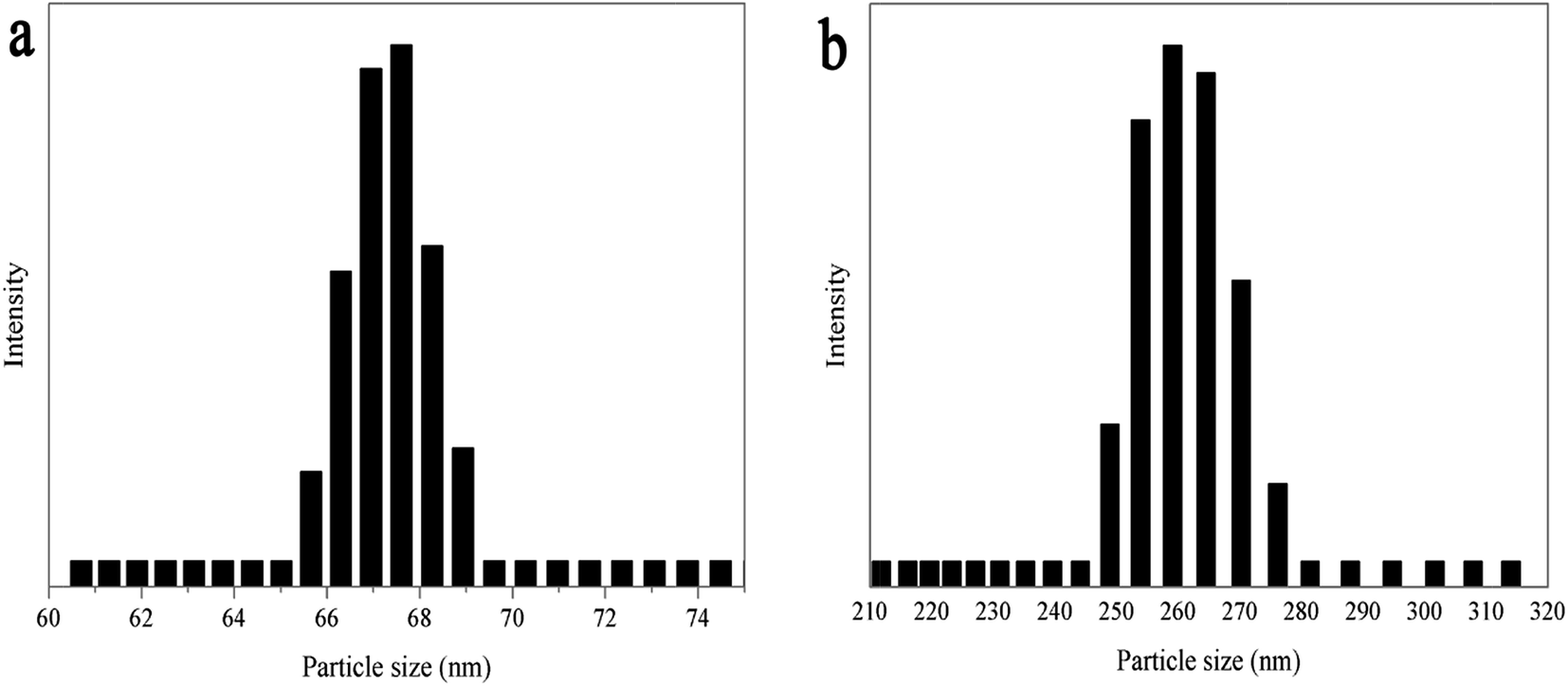
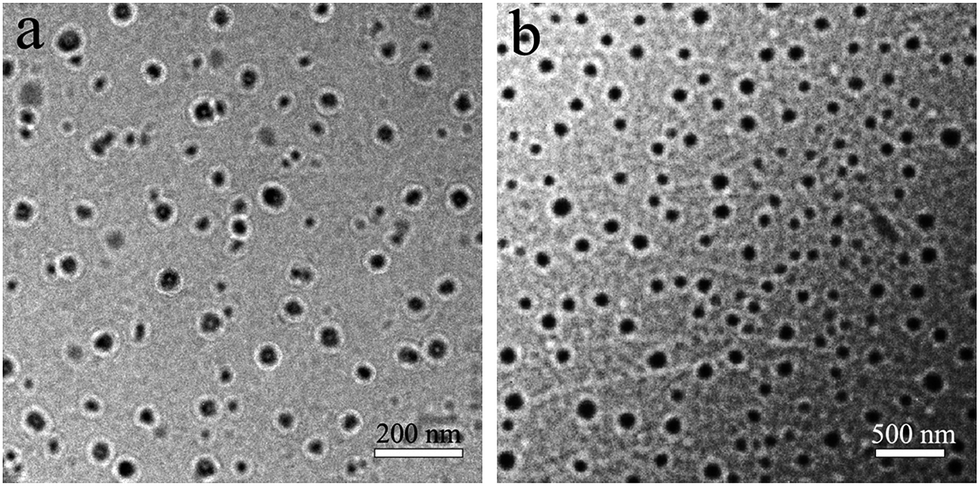
As shown in Fig. 4, the particle size and size distribution of pure WPU and WPU/h-NZFO-1.0 emulsions were tested by dynamic light scattering (DLS). It was observed that the average particle size of pure WPU emulsion (Fig. 4a) is only 67.2 nm with narrow distribution width. The average particle size of WPU/h-NZFO-1.5 nanocomposite emulsion (Fig. 4b) is around 251.8 nm. Therefore, the introduction of h-NZFO-VTES nanoparticles increased the particle size of nanocomposite. The structure and morphology of pure WPU and WPU/h-NZFO-1.5 emulsions were identified by TEM. From Fig. 5, we can see that the WPU particles (Fig. 5a) are well dispersed with very narrow distribution. The h-NZFO-VTES nanoparticles in Fig. 5b are wrapped in WPU matrix without large aggregation. Meanwhile, the emulsion became darker and the particle size increased when the h-NZFO-VTES was introduced, consistent with above DLS results. Moreover, the particle size of both pure WPU and WPU/h-NZFO-1.5 obtained from TEM were smaller than the results observed from DLS because of dehydration process. The introduction of h-NZFO-VTES led to more hydrophobic structure and improvement in the cross-linking density of WPU polymers. Consequently it is difficult to emulsify the waterborne polyurethane with a constant –COOH content in the polymer chain and the particle size increases.37
 |
| | Fig. 4 Particle size and distribution of WPU (a) and the WPU/h-NZFO-1.5 (b) emulsions. | |
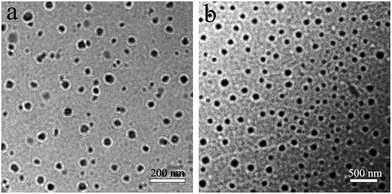 |
| | Fig. 5 TEM micrographs of pure WPU (a) and the WPU/h-NZFO-1.5 (b) emulsions. | |
3.4 Properties characterization of WPU/h-NZFO nanocomposites
3.4.1 Effect of h-NZFO-VTES on stability of WPU/h-NZFO emulsions. Generally, WPU emulsion is stored for a certain time prior to use, therefore, storage stability is an important characteristic of polymer emulsion. The storage stability of WPU/h-NZFO emulsions with different h-NZFO-VTES content was decided by stable time. The longer stable time indicates a better emulsion stability. Table 2 shows the results of emulsion stability. It was observed that when the h-NZFO-VTES content was lower than 1.5%, the WPU/h-NZFO emulsions can be preserved longer than 43 days, which is higher than that of some reported literatures.38,39 The h-NZFO nanoparticles prepared in this paper show smaller diameter and better dispersion than that of previous report, and this facilitates the improvement in emulsion stability. However, the storage stability decreased with the increasing of h-NZFO-VTES content; the stable time reduced to 34 days for WPU/h-NZFO-2.0 (still longer than WPU/h-NZFO-2.0 of 30 days prepared in previous work) and 17 days for WPU/h-NZFO-3.0 emulsion. The effect of h-NZFO-VTES content on storage stability can be explained that the h-NZFO-VTES particles were well wrapped and dispersed in WPU matrix at low h-NZFO-VTES concentration. At high h-NZFO-VTES content, the h-NZFO-VTES (especially the unreactive particles) were more likely to gather together to form large aggregates, which easily separated from the organic matrix system, leading to poor emulsion stability.
Table 2 Storage stability of WPU/h-NZFO nanocomposite emulsions with different h-NZFO-VTES contents
| h-NZFO-VTES content (wt%) |
0.5 |
1.0 |
1.5 |
2.0 |
3.0 |
| Stable time/day |
43 |
43 |
43 |
34 |
17 |
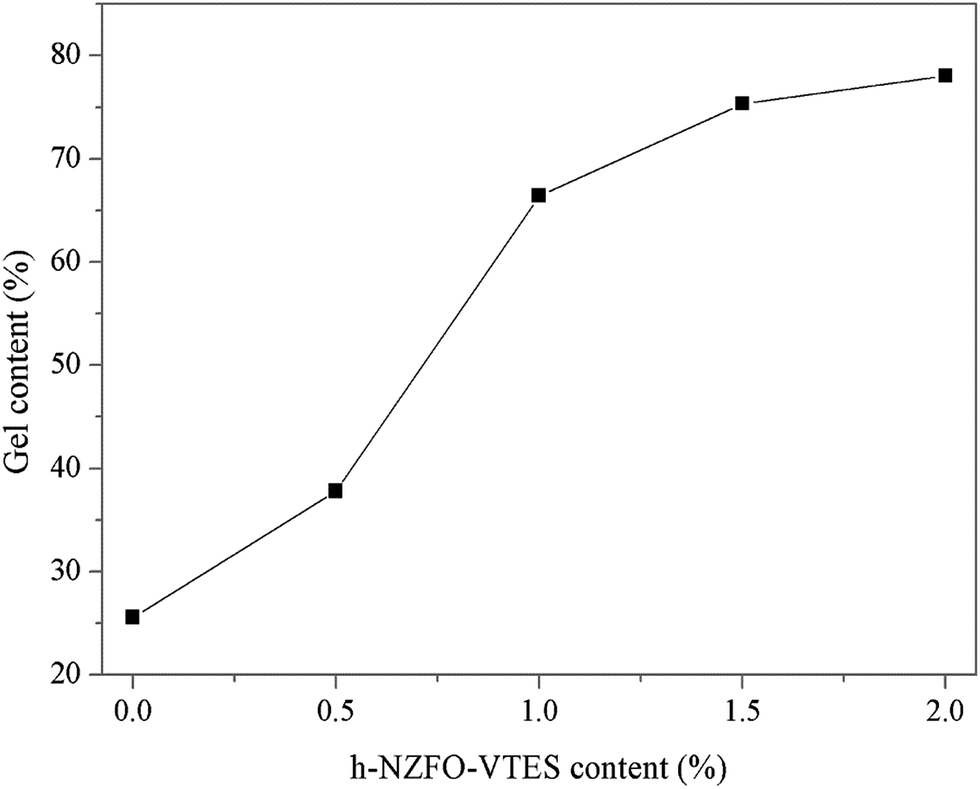
3.4.2 Gel content of WPU/h-NZFO nanocomposites. Gel content is an important parameter regarding the degree of cure and cross-linking density of polymers. As can be seen from Fig. 6, the gel content of film materials considerably increased as the h-NZFO-VTES content increased. The gel content reached up to 78.1% when the h-NZFO-VTES loading was 2.0%. This should be attributed to the polymerization reaction. The functionalized h-NZFO-VTES particles contain numerous CC bonds, and they acted as new crosslinker in emulsions can be cross-linked with WPU pre-polymers during the UV curing process, leading to the formation of network structure, which would result the improvement in cross-linking density of WPU/h-NZFO nanocomposites. Therefore, the gel content increased as h-NZFO-VTES content increased.
 |
| | Fig. 6 Influence of h-NZFO-VTES content on the gel content of the WPU nanocomposites. | |
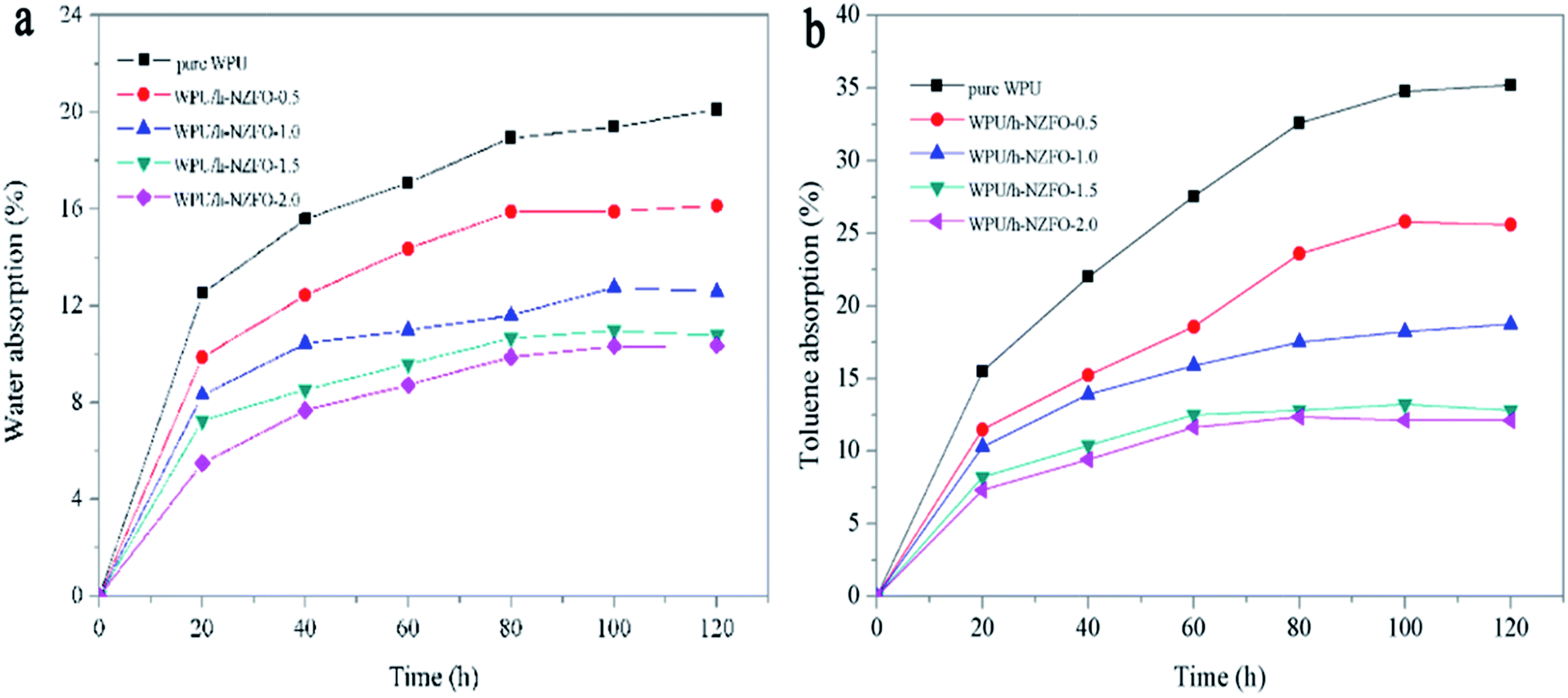
3.4.3 Water and toluene absorption of WPU/h-NZFO nanocomposite films. Fig. 7a and b exhibit the water and toluene absorption of pure WPU and WPU/h-NZFO nanocomposite films with different h-NZFO-VTES contents, respectively. As the time went on, the water absorption of all samples increased firstly and reached a balance after 100 h. The water absorption of pure WPU is 19.40% in 100 h. Generally speaking, the water absorption of WPU/h-NZFO nanocomposites decreased with the increasing of h-NZFO-VTES loading. The water absorptions of WPU/h-NZFO nanocomposites with h-NZFO-VTES content are 0.5%, 1.0%, 1.5%, and 2.0% are 15.89%, 12.76%, 10.98%, and 10.32% in 100 h, respectively. Namely the introduction of h-NZFO-VTES particles improved water resistance of the nanocomposites. This may be attributed to the formation of cross-linking structure in WPU/h-NZFO films. As can be seen from Fig. 7b, the toluene absorption of WPU/h-NZFO nanocomposite films decreased with the increasing of h-NZFO-VTES loading. The toluene absorption for pure WPU is 34.76% in 100 h while reduced to 12.11% for WPU/h-NZFO-2.0 film. The reason for this as above stated for water absorption. In summary, the more h-NZFO-VTES the composites contain, the better water and toluene resistance the WPU/h-NZFO films have.
 |
| | Fig. 7 Water absorption (a) and toluene absorption (b) of pure WPU and the WPU/h-NZFO nanocomposites. | |
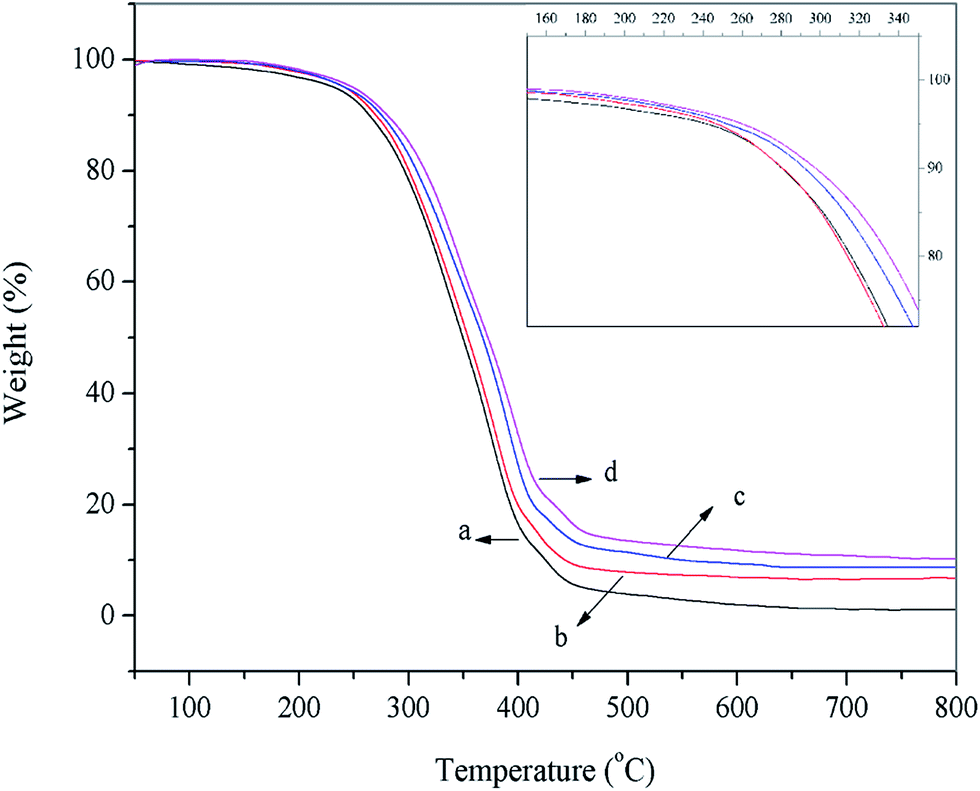
3.4.4 Thermal behavior of WPU/h-NZFO nanocomposite films. TGA test was conducted to investigate the effect of h-NZFO-VTES content on the thermal stability of the WPU/h-NZFO nanocomposites. Fig. 8 presented the typical TGA curves of pure WPU and WPU/h-NZFO films. It was found that the h-NZFO-VTES content was greatly affected by the thermal properties of the nanocomposite films. TGA curves of all samples displayed two distinct degradation stages. The first weigh loss stage, from 228 to 410 °C, is possibly attributed to the degradation of the hard segment of the WPU matrix. The second weigh loss occurred between 410 and 562 °C, which is due to the degradation of the soft segment of the WPU polymers.10,40 The initial degradation temperature increased from 241 °C to 288 °C for WPU/h-NZFO-2.0 (Fig. 8d). Meanwhile, the thermostability also increased with h-NZFO-VTES loading increased from 2.67% to 13.21%. The reason might be that the strong interaction between h-NZFO-VTES and WPU matrix due to the formation of cross-linking structure in WPU/h-NZFO. On the other hand, the excellent thermal stability of h-NZFO-VTES inorganic particles also contributed to the thermal stability of films. These results implied that the addition of h-NZFO-VTES enhanced the thermal stability of WPU/h-NZFO films.
 |
| | Fig. 8 TGA curves of pure WPU (a), WPU/h-NZFO-0.5 (b), WPU/h-NZFO-1.5 (c), and WPU/h-NZFO-2.0 (d) nanocomposites. | |
To further investigate the thermal mechanical properties of film samples, the dynamic mechanical thermal analysis (DMA) was performed. Fig. 9 show the loss factor (tanδ) and the storage modulus (E′) curves of pure WPU and WPU/h-NZFO films with different h-NZFO-VTES loading, respectively. The temperature corresponded to the highest point of the tanδ curve was defined as the glass transition temperature (Tg). The Tg of pure WPU (Fig. 9a) is 52.1 °C while individually increased to 70.3, 88.2, and 101.6 °C for WPU/h-NZFO-0.5, WPU/h-NZFO-1.0, and WPU/h-NZFO-2.0 films, respectively. This implied that the increasing h-NZFO-VTES content has a positive effect on Tg value of WPU/h-NZFO nanocomposites. This could be attributed to the strong interaction between inorganic and organic phases on one hand. When the phase mixing was relatively complete in WPU/h-NZFO nanocomposites, the movement of chain segments was greatly restricted. On the other hand, inorganic nanoparticles usually hinder the motion of polymer chains, also leading to an increase in the peak temperature of the nanocomposite.12 Besides, the improvement in cross-linking density made the backbone mobility difficult, resulting in the increase of Tg value. It was observed from Fig. 9b that the storage modulus increased with increasing h-NZFO-VTES loading. For inorganic/polymer composites, the storage modulus depend on the inorganic phase and cross-linking density. The incorporation of inorganic particles generally enhanced the storage modulus. In this system, the increased cross-linking density of nanocomposites should be responsible for the enhancement of the storage modulus.
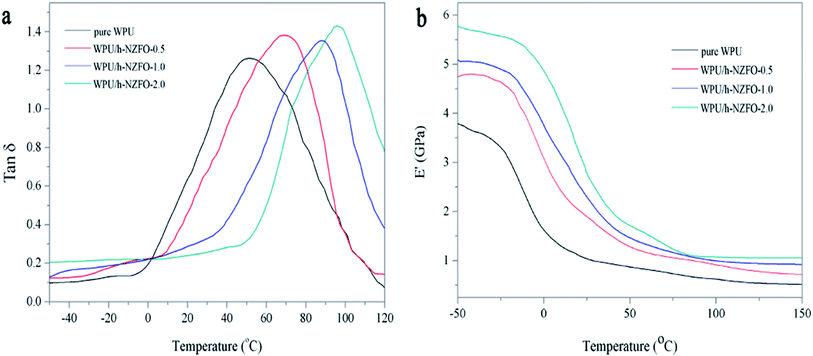 |
| | Fig. 9 The loss factor (a) and storage modulus curves (b) of pure WPU and the WPU/h-NZFO nanocomposites. | |
3.4.5 Magnetic properties of h-NZFO and WPU/h-NZFO nanocomposites. The magnetic properties of samples were investigated using VSM at room temperature. Fig. 10 shows the magnetic hysteresis loops of h-NZFO and h-NZFO-VTES. The pristine h-NZFO achieved a high saturation magnetization value (Ms) of 81.78 emu g−1. Because of the existence of organic groups on the surface of h-NZFO, the saturation magnetization reduced to 75.88 emu g−1 of h-NZFO-VTES. It can be explained that the VTES had effectively grafted to the h-NZFO surface. Fig. 11 shows the magnetic hysteresis loops of WPU/h-NZFO nanocomposite films with different h-NZFO-VTES loading. It was observed that the saturation magnetization for all WPU/h-NZFO samples were sharply decreased compared to the pure h-NZFO-VTES particles and exhibited better magnetism. The saturation magnetization of WPU/h-NZFO films increased as h-NZFO-VTES content increased. The saturation magnetization of WPU/h-NZFO-1.0 is 11.45 emu cm−3, which is much higher than WPU/unmodified h-NZFO-1.0 (5.98 emu cm−3). The magnetism of nanocomposites is greatly affected by nanoparticle size, surface disorder and nanoparticle distribution.41 The functionalization of h-NZFO nanoparticles improved the dispersion of h-NZFO nanoparticles in the WPU matrix, which will contribute to excellent magnetism. Furthermore, the saturation magnetization of WPU/h-NZFO-2.0 reached up to 24.2 emu cm−3, this is higher than some reported film materials.42–44 The WPU/h-NZFO nanocomposites have obtained magnetism from h-NZFO-VTES magnetic nanoparticles and displayed superior magnetic properties, and these materials would have a potential application prospect in microwave absorption fields.
 |
| | Fig. 10 Magnetic hysteresis loops of pristine h-NZFO and h-NZFO-VTES. | |
 |
| | Fig. 11 Magnetic hysteresis loops of WPU/h-NZFO nanocomposites with different h-NZFO-VTES contents. | |
3.4.6 Mechanical properties of pure WPU and WPU/h-NZFO nanocomposites. The influence of h-NZFO-VTES loading on mechanical properties of WPU/h-NZFO films was investigated using the computer controlled universal tensile testing machine. Table 3 exhibits the tensile strength and elongation at break of pure WPU and WPU/h-NZFO nanocomposites. It was found that both the tensile strength and elongation at break were greatly affected by h-NZFO-VTES content. As shown in Table 3, the pure WPU presented relatively lower tensile strength of 19.70 MPa and higher elongation at break of 397.63%. The tensile strength of WPU/h-NZFO nanocomposite films increased as the h-NZFO-VTES content increased and reached the maximum value of 47.89 MPa with h-NZFO-VTES content of 2.0%. This indicated that the introduction of h-NZFO-VTES particles enhanced tensile properties of materials. However, the elongation at break showed a downward trend relative to pure WPU. The increase of tensile strength could be attributed to the strong chemical interaction formed between h-NZFO-VTES and WPU matrix, because the inorganic particles combined with the WPU polymers through covalent bond during the curing process. What's more, the improvement in cross-linking density of materials should be also responsible for the decrease in the elongation at break of WPU/h-NZFO nanocomposites.
Table 3 Influence of h-NZFO-VTES content on mechanical properties of WPU/h-NZFO nanocomposites
| h-NZFO-VTES content (wt%) |
Tensile strength/MPa |
Elongation at break (%) |
| 0 |
19.70 |
397.63 |
| 0.5 |
26.32 |
339.37 |
| 1.0 |
34.90 |
306.48 |
| 1.5 |
45.84 |
272.43 |
| 2.0 |
47.89 |
254.18 |
4. Conclusion
A series of WPU/h-NZFO nanocomposite films containing 0, 0.5, 1.0, 1.5, and 2.0 wt% content of h-NZFO-VTES were successfully prepared by in situ polymerization method. The morphology of WPU/h-NZFO films was characterized and suggested that h-NZFO-VTES particles were homogeneously dispersed in WPU matrix. Meanwhile, the introduction of h-NZFO-VTES obviously improved the gel content, thermal stability, and tensile strength of materials. Furthermore, the VSM test indicated that the resulting WPU/h-NZFO films possessed superior magnetic properties at lower h-NZFO-VTES loading. In summary, this work provided an alternative method to prepare environmentally friendly WPU/h-NZFO microwave absorption materials. Further investigation is now in progress.
Acknowledgements
The authors would like to acknowledge the financial support provided by the National Natural Science Foundation of China (No. 21301081), the Natural Science Foundation of Gansu Province (1506RJYA221), and the Fundamental Research Funds for the Central Universities (lzujbky-2015-30).
References
- N. E. Marcovich, M. L. Auad, N. E. Bellesi, S. R. Nutt and M. I. Aranguren, J. Mater. Res., 2006, 21, 870–881 CrossRef CAS.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- J. Ramos, A. Millan and F. Palacio, Polymer, 2011, 41, 8461–8464 CrossRef.
- P. Xu, X. Han, C. Wang, H. Zhao, J. Wang, X. Wang and B. Zhang, J. Phys. Chem. B, 2008, 112, 2775–2781 CrossRef CAS PubMed.
- L. Zhai, R. Liu, F. Peng, Y. Zhang, K. Zhong, J. Yuan and Y. Lan, J. Appl. Polym. Sci., 2013, 128, 1715–1724 CrossRef CAS.
- W. Zhou, X. Hu, X. Bai, S. Zhou, C. Sun, J. Yan and P. Chen, ACS Appl. Mater. Interfaces, 2011, 3, 3839–3845 CAS.
- G. Liao, S. Chen, X. Quan, H. Chen and Y. Zhang, Environ. Sci. Technol., 2010, 44, 3481–3485 CrossRef CAS PubMed.
- S. Liu, H. Xu, J. Ou, Z. Li, S. Yang and J. Wang, Mater. Chem. Phys., 2012, 132, 500–504 CrossRef CAS.
- M. Jouni, A. Boudenne, G. Boiteux, V. Massardier, B. Garnier and A. Serghei, Polym. Compos., 2013, 34, 778–786 CrossRef CAS.
- Z. Spitalsky, D. Tasis, K. Papagelis and C. Galiotis, Prog. Polym. Sci., 2010, 35, 357–401 CrossRef CAS.
- L. Peng, L. Zhou, Y. Li, F. Pan and S. Zhang, Compos. Sci. Technol., 2011, 71, 1280–1285 CrossRef CAS.
- A. Maity and M. Biswas, J. Ind. Eng. Chem., 2006, 12, 311–351 CAS.
- R. K. Bharadwaj, Macromolecules, 2001, 34, 9189–9192 CrossRef CAS.
- N. Nabih, K. Landfester and A. Taden, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5019–5029 CrossRef CAS.
- D. Wang, J. He, N. Rosenzweig and Z. Rosenzweig, Nano Lett., 2004, 4, 409–413 CrossRef CAS.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S. W. Tsang, T. H. Lai and F. So, Nat. Photonics, 2012, 6, 115–120 CrossRef CAS.
- F. N. Crespilho, R. M. Iost, S. A. Travain, O. N. Oliveira and V. Zucolotto, Biosens. Bioelectron., 2009, 24, 3073–3077 CrossRef CAS PubMed.
- S. H. Hsu, C. W. Chou and S. M. Tseng, Macromol. Mater. Eng., 2004, 289, 1096–1101 CrossRef CAS.
- J. Huybrechts, P. Bruylants, A. Vaes and A. De Marre, Prog. Org. Coat., 2000, 38, 67–77 CrossRef CAS.
- H. Sardon, L. Irusta, M. J. Fernández-Berridi, M. Lansalot and E. Bourgeat-Lami, Polymer, 2010, 51, 5051–5057 CrossRef CAS.
- C. C. Santos, M. C. Delpech and F. M. Coutinho, J. Mater. Sci., 2009, 44, 1317–1323 CrossRef CAS.
- M. S. Yen and P. Y. Tsai, J. Appl. Polym. Sci., 2003, 90, 233–243 CrossRef CAS.
- J. J. Chen, C. F. Zhu, H. T. Deng, Z. N. Qin and Y. Q. Bai, J. Polym. Res., 2009, 16, 375–380 CrossRef.
- I. S. Gunes, F. Cao and S. C. Jana, Polymer, 2008, 49, 2223–2234 CrossRef CAS.
- S. Subramani, J. Y. Lee, S. W. Choi and J. H. Kim, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2747–2761 CrossRef CAS.
- D. Molina, G. Griffini, M. Levi and S. Turri, Polym. Adv. Technol., 2014, 25, 1082–1088 CrossRef CAS.
- R. O. N. G. Min Zhi and Q. Zhang, Polym. J., 2006, 38, 799–806 CrossRef.
- S. Zhang, J. Jiang, C. Yang, M. Chen and X. Liu, Prog. Org. Coat., 2011, 70, 1–8 CrossRef CAS.
- P. Gao, E. V. Rebrov, T. M. Verhoeven, J. C. Schouten, R. Kleismit, G. Kozlowski and G. Subramanyam, J. Appl. Phys., 2010, 107, 044317 CrossRef.
- D. L. Zhao, Q. Lv and Z. M. Shen, J. Alloys Compd., 2009, 480, 634–638 CrossRef CAS.
- A. Khan and P. G. Smirniotis, J. Mol. Catal. A: Chem., 2008, 280, 43–51 CrossRef CAS.
- E. Açıkalın, O. Atıcı, A. Sayıntı, K. Çoban and H. Erkalfa, Prog. Org. Coat., 2013, 76, 972–978 CrossRef.
- Q. Li, Y. Li, X. Li, S. Chen, S. Zhang, J. Wang and C. Hou, J. Alloys Compd., 2014, 608, 35–43 CrossRef CAS.
- F. Yan, J. Li, J. Zhang, F. Liu and W. Yang, J. Nanopart. Res., 2009, 11, 289–296 CrossRef CAS.
- B. Das, P. Chattopadhyay, D. Mishra, T. K. Maiti, S. Maji, R. Narayan and N. Karak, J. Mater. Chem. B, 2013, 1, 4115–4126 RSC.
- W. Wang, S. P. Gumfekar, Q. Jiao and B. Zhao, J. Mater. Chem. C, 2013, 1, 2851–2859 RSC.
- P. Calvo, C. Remunan-Lopez, J. L. Vila-Jato and M. J. Alonso, J. Appl. Polym. Sci., 1997, 63, 125–132 CrossRef CAS.
- L. Zhang, H. Zhang and J. Guo, Ind. Eng. Chem. Res., 2012, 51, 8434–8441 CrossRef CAS.
- S. Zhang, Y. Li, L. Peng, Q. Li, S. Chen and K. Hou, Composites, Part A, 2013, 55, 94–101 CrossRef CAS.
- Z. Dai, Z. Li, L. Li and G. Xu, Polym. Adv. Technol., 2011, 22, 1905–1911 CrossRef CAS.
- Z. Huang and F. Tang, J. Colloid Interface Sci., 2004, 275, 142–147 CrossRef CAS PubMed.
- M. Ashjari, A. R. Mahdavian, N. G. Ebrahimi and Y. Mosleh, J. Inorg. Organomet. Polym. Mater., 2010, 20, 213–219 CrossRef CAS.
- S. H. Ahmad, M. H. Abdullah, D. Hui, A. N. Yusoff and D. Puryanti, J. Magn. Magn. Mater., 2010, 322, 3401–3409 CrossRef.
- J. O. Park, K. Y. Rhee and S. J. Park, Appl. Surf. Sci., 2010, 256, 6945–6950 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.