
Efficient copolymerization of ethylene with norbornene or its derivatives using half-metallocene zirconium(iv) catalysts†
Abstract
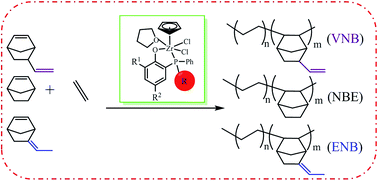
A series of half-metallocene zirconium complexes CpZr(thf)Cl2[O-2,4-tBu-6-(P-PhR)C6H2] (Cp = C5H5, 2a: R = Me, 2b: R = tBu, 2c: R = Ph, 2d: R = 4-F-Ph) have been synthesized by reacting CpZrCl3 with the corresponding ligands in THF. All the complexes were characterized by (1H, 13C and 31P) NMR spectroscopy as well as elemental analysis. X-ray structural analysis for 2a and 2d revealed a six-coordinate distorted octahedral geometry around the zirconium center in the solid state. With the activation of a suitable cocatalyst, half-metallocene zirconium complexes 2a–d were employed to catalyze ethylene polymerization and ethylene copolymerization with cycloolefins including norbornene (NBE), 5-ethylidene-2-norbornene (ENB) and 5-vinyl-2-norbornene (VNB). All the complexes exhibited high efficiency toward the copolymerizations. High comonomer incorporations up to 62% (NBE), 76% (ENB) and 40.6% (VNB), respectively, with high catalytic activities above 103 kgpolymer per molZr per h were achieved using catalyst 2c. Steric hindrance and electronic effects of the complexes or the comonomers greatly influence copolymerization behavior. Thus, catalytic activity and copolymer chain structure, including comonomer incorporation and the molecular weight, can be easily tuned over a wide range by changing catalyst and reaction conditions.
 Please wait while we load your content...
Please wait while we load your content...

