
Fabrication of magnetite nanocrystals in alcohol/water mixed solvents: catalytic and colloid property evaluation†
Abstract
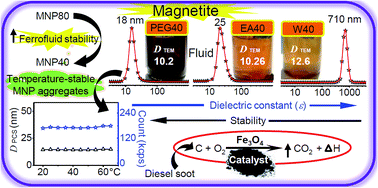
In this work, Fe3O4 nanocrystals have been synthesized by homogeneous precipitation in different alcohol/water (1 : 1) solvent mixtures at two different temperatures to elucidate the role of the dielectric constant (ε) of the reaction medium. The effects of different solvents on the catalytic activity of precipitated NPs in carbon combustion were examined. HRTEM images, SAED and XRD confirmed that the nanocrystals are of pure fcc inverse spinel Fe3O4 phase with narrow size distribution, and the crystals are completely dispersible in water. The morphological features of the nanocrystals, such as their surface termination and shape of the Fe3O4 NPs, were analyzed by HR-TEM. As ε decreases, the crystal size decreases for mono-ol systems compared to ∼13 nm size in water, whereas ethylene glycol/water yields finer ∼8.2 nm crystals although it has the highest ε among the mono-/poly-ols. A soot combustion study demonstrates that the catalytic activity is mainly due to the available surface area along with the exposure of active crystallographic facets. A study of the colloids by light scattering shows that the alcohol mediated process produces 16 to 33 nm MNP clusters composed of 2 to 3 particles in highly stable aqueous magnetic fluids. The relatively high temperature process favors higher crystallinity and particle size with reduced colloidal stability in the aqueous phase. The nanocrystalline powders and the dispersed colloids have excellent potential applications in biotechnology and selective catalysis and also as ferrofluids.
 Please wait while we load your content...
Please wait while we load your content...

